

Volume 9, Issue 4 (11-2022)
JROS 2022, 9(4): 187-196 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Shams R, Zabihiyeganeh M, Aminian A, Sarikhani M. Advancements in Understanding and Managing Hypophosphatasia: From Clinical Spectrum and Diagnosis to Therapeutic Strategies. JROS 2022; 9 (4) :187-196
URL: http://jros.iums.ac.ir/article-1-2232-en.html
URL: http://jros.iums.ac.ir/article-1-2232-en.html



1- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran.
Full-Text [PDF 602 kb]
(156 Downloads)
| Abstract (HTML) (546 Views)
Full-Text: (206 Views)
1. Introduction
In 1948, John C. Rathbun first identified a condition characterized by rickets, seizures, and diminished levels of alkaline phosphatase (AP) in a patient, a condition later known as hypophosphatasia (HPP) [1]. HPP emerges due to mutations affecting the gene for the tissue-nonspecific isoenzyme of AP (TNSALP), presenting a broad spectrum of systemic metabolic disturbances [2]. The AP-liver (ALPL) gene, responsible for HPP, is predominantly active in the bones, liver, kidneys, and developing teeth, still, its expression can also be found in the central nervous system, fibroblasts, endothelial cells, and several other cell types [3]. Despite its rarity, HPP is a significant yet often overlooked cause of osteomalacia and susceptibility to atypical fractures in adults, leading to a critical need for heightened awareness among healthcare professionals. This awareness is crucial to avoid the inappropriate use of antiresorptive therapies, such as bisphosphonates and denosumab, in these patients. Such treatments, intended for osteoporosis, can heighten the risk of atypical femoral fractures (AFF). The overarching goal of this comprehensive review is not only to enhance awareness and understanding among healthcare professionals, particularly in orthopedics and rheumatology but also to lay the groundwork for the development of evidence-based clinical practices. By consolidating recent advances and highlighting the necessity for appropriate therapeutic approaches, this study endeavors to improve patient outcomes and prevent the mismanagement of this complex condition.
2. Genetics and Molecular Pathophysiology
The molecular pathophysiology and pathogenesis of HPP are primarily centered on mutations in the ALPL gene (MIM*171760), which encodes the TNSALP, UniprotKB# P05186). This enzyme is crucial for bone and dental mineralization, and its dysfunction leads to the clinical manifestations of HPP. HPP results from mutations in the ALPL gene located on the short arm of chromosome 1 (1p36.1-34) [4]. These mutations can be heterozygous, compound heterozygous, or homozygous, with over 400 different mutations identified, including missense/nonsense mutations, deletions, and insertions [5]. A recent investigation analyzed the genetic inheritance patterns within a group of 424 individuals diagnosed with HPP. The results of this study indicated that the genetic characteristics of this cohort can be classified into three distinct clinical categories, severe HPP, which is both recessive and infrequent; moderate HPP, occurring more frequently and inheritable as either recessive or dominant; and mild HPP, the most common form, predominantly inherited in a dominant manner, marked by decreased levels of AP and nonspecific clinical symptoms [6]. However, mutations can lead to impaired function of the enzyme produced by TNSALP. This inborn metabolic disorder causes the extracellular buildup of natural substrates of TNSALP, including phosphoethanolamine (PEA), an integral part of the phosphatidylinositol-glycan linkage apparatus linking APs and other proteins to plasma membrane surfaces. Another substrate is inorganic pyrophosphate (PPi), recognized for hindering hydroxyapatite crystal formation and impacting biomineralization. Additionally, pyridoxal 5′-phosphate (PLP), the primary circulating form of vitamin B6 (B6), accumulates [7]. TNSALP is a glycosylphosphatidylinositol-anchored membrane enzyme expressed in various tissues, including bone, liver, kidneys, and teeth. It is involved in the dephosphorylation of several substrates, such as inorganic PPi, a natural inhibitor of hydroxyapatite crystal formation and growth [8]. PPi plays a pivotal role in regulating bone mineralization. It prevents excessive crystal growth, ensuring proper bone formation and remodeling. TNSALP’s activity is essential for maintaining the balance between PPi and phosphate (Pi) levels, facilitating normal bone mineralization [8]. Mutations in the ALPL gene reduce TNSALP activity, leading to the accumulation of PPi. High levels of PPi inhibit the deposition of calcium and Pi into the bone matrix, causing hypomineralization of bone and teeth, the hallmark of HPP. The mineralization defect results in a spectrum of skeletal abnormalities, ranging from severe perinatal forms with almost no bone mineralization to milder adult forms characterized by recurrent fractures, osteomalacia, and dental issues [9].
3. Clinical Presentations
HPP displays a diverse clinical spectrum, encompassing severe manifestations in infants, including life-threatening conditions and even fetal death, to milder forms observed in adults, characterized by dental or skeletal symptoms [10]. The classification of HPP involves six distinct forms, determined by factors, such as the age of onset, the severity of symptoms, and the specific clinical manifestations exhibited by individuals [3]. This classification system aids in understanding the varied presentations of HPP across different age groups and the severity of the condition.
Benign prenatal HPP
The prenatal benign variant of HPP is identifiable through gestational ultrasonography, revealing characteristics, such as shortened long bones and hypomineralization [1]. These manifestations often exhibit spontaneous improvement during late pregnancy or postnatal development. The natural progression of this form is unpredictable, with the potential for evolving from odonto HPP to the more severe infantile form [3]. The variability in its natural course underscores the challenge of foreseeing the trajectory of this prenatal benign form.
Perinatal (sever) HPP
This form, recognized as the most critical form, is primarily inherited through autosomal recessive traits and is considered rare. The clinical manifestations typically become evident by the end of pregnancy or at birth [3, 11], with a prognosis that is unfortunately almost always fatal shortly thereafter [10]. Radiological examinations reveal characteristic changes, including bone hypomineralization, radiolucent regions resembling “tongues” at the ends of long bones, slender and deformed bones, metaphyses presenting a “moth-eaten” and cup-shaped appearance, and the presence of osteochondral spurs [12]. Additional features encompass a high-pitched cry, pyridoxine-dependent seizures, periodic apnea accompanied by cyanosis and bradycardia, irritability, unexplained fever, myelophthisic anemia, and intracranial hemorrhage [10]. The serum ALP levels in neonates with this form are typically excessively low or undetectable [3]. Research results indicate that most mutations associated with this particular subtype are missense mutations. Establishing a correlation between genotype and phenotype can contribute to the development of a novel classification system. Such a classification of ALPL variants holds the potential to enhance the accuracy of distinguishing between perinatal lethal cases and other types of HPP [13, 14].
Infantile HPP
This form of HPP represents a moderately severe phenotype and is commonly associated with autosomal recessive inheritance [3]. Although newborns affected by this type may seem healthy at birth, clinical manifestations typically become apparent within the initial 6 months of life [15]. As mentioned, HPP causes the extracellular buildup of natural substrates of TNSALP, including PEA, PPi, and PLP. An established complication arises in life-threatening HPP during infancy due to compromised dephosphorylation of PLP to pyridoxal (PL) by TNSALP. PL, a B6 vitamer capable of crossing membranes, is then rephosphorylated to PLP or pyridoxamine Pi for intracellular enzymatic reactions. This compromised dephosphorylation process can result in B6-dependent seizures in infancy [16]. On the other hand, the increased excretion of calcium may contribute to renal damage. Despite the presence of an open fontanelle, premature craniosynostosis is frequently observed, potentially leading to elevated intracranial pressure [15]. Historically, it has been estimated that around 50% of infants with this subtype do not survive infancy [17]. A poor prognosis is often associated with the presence of respiratory failures, such as rib fractures, thoracic deformity, recurrent pneumonias, and or pyridoxine-dependent seizures [3].
Childhood HPP
Childhood HPP represents a highly heterogeneous clinical entity within the broader spectrum of HPP [15], exhibiting a notable diversity further categorized into mild and severe forms. In instances of mild presentation, affected children typically demonstrate good overall health, normal physical functionality, and minimal or negligible symptoms [18]. However, early-onset tooth loss is a characteristic feature, accompanied by subtle skeletal alterations, such as diminished bone mass evident in radiographic assessments. Conversely, severe childhood HPP, typically inherited as an autosomal recessive trait, manifests more prominently with challenges [19]. Patients experience premature tooth loss concomitant with skeletal pain, precipitating episodes of unexplained crying, and reluctance to ambulate. This muscular weakness contributes to delayed ambulation, a characteristic waddling gait, and difficulties in ascending stairs. Rachitic deformities may manifest as cranial misshaping, costochondral junction beading, bowed or knock-kneed legs, and joint enlargement due to metaphyseal flaring. While spontaneous remission of bone disease is acknowledged, the condition may reoccur during middle or late adulthood [20].
Adult HPP
The manifestation of HPP in adults emerges in middle age, with diagnoses occurring in individuals aged 18 years or older [15]. However, it is noteworthy that many adults with HPP have histories of signs or symptoms preceding this age that were not recognized during childhood, leading to significant delays in diagnosis [21]. Approximately 40%–55% of adults with HPP have a documented history of fractures, often multiple. At presentation, nearly two-thirds of patients exhibit symptoms, with musculoskeletal pain being a prevalent complaint (40%–75% in various series), affecting diverse areas, such as the feet, ankles, knees, thighs, hips, back, and joints [22]. Fractures commonly occur in the feet and femur/hip, but also in the wrists, vertebrae, or other bones, and are characterized by slow healing and potential recurrence. Bone mineral density (BMD) can vary, being low, normal, or high, with high lumbar spine BMD paradoxically associated with a higher risk of fractures. Dentition is also affected, with early loss of adult teeth being a common feature (25–35%) [2, 3]. Osteomalacia and pseudofractures, particularly in the femur, may precede HPP diagnosis, leading to bowing deformities of long bones in about 15% of patients. Radiographic calcific periarthritis, chondrocalcinosis, ossification of ligaments, scoliosis, and other symptoms, such as headaches, chronic fatigue, and gait abnormalities are observed in varying percentages [23]. Adult HPP, especially mild forms, may remain asymptomatic, contributing to underdiagnosis, as symptoms like musculoskeletal pain are non-specific and prevalent in the population. A study recently highlighted the clinical presentation and genetic profile of HPP in a cohort of 19 Chinese adults. The participants’ median age was 62, ranging from 32 to 74 years, with a predominance of female patients (16 out of 19). The most frequently reported issues were musculoskeletal symptoms (63.2%), dental complications (42.1%), fractures (36.8%), and fatigue (31.6%). Nearly half of the patients (47.4%) were incorrectly diagnosed with osteoporosis, and a third of them had undergone treatment with anti-resorptive drugs. The average serum ALP level among the group was notably low at 29.1 U/L, with 94.7% (18 out of 19) displaying ALP levels beneath the 40 U/L threshold. Genetic testing unveiled 14 different mutations in the ALPL gene, three of which were previously unreported [24]. Therefore, the diagnostic challenge arises in distinguishing between adult HPP and a normal phenotype, particularly in patients heterozygous for an ALPL gene mutation. The mentioned study has reported that the symptoms of two patients with compound heterozygous mutations were more severe than those with heterozygous mutations. Debilitating consequences of adult HPP include recurrent fractures, skeletal and joint pain, and muscle weakness. Notably, AFF can occur in up to 10% of HPP patients, with a higher frequency observed in those exposed to antiresorptive drugs before AFF onset [25, 26].
Odonto HPP
OdontoHPP is identified as the least severe yet most prevalent phenotype of HPP, affecting individuals across a broad age spectrum, encompassing both pediatric and adult populations [15]. This variant is primarily distinguished by its dental manifestations, which occur in the absence of radiographic or histopathological evidence indicative of rickets or osteomalacia. For instance, a recent study documented the case of a 2-year-old Japanese child diagnosed with odontoHPP. The child experienced premature loss of deciduous teeth, with the roots remaining intact. Serum analysis revealed low levels of AP alongside significantly elevated levels of PEA and PLP. Genetic testing of the ALPL gene identified a heterozygous mutation (NM_000478.6:c.1151C > A, p.Thr384Lys) [27]. Clinical presentations of these patients include the asymptomatic, premature loss of primary incisors, where the dental roots remain intact, and this occurs without accompanying gingival inflammation, ulceration, dental abscess formation, or a documented history of trauma. In the diagnostic assessment of patients presenting with early onset (before age 5 years) of unexplained tooth loss or the presence of abnormally loose teeth upon dental examination, odontoHPP should be considered as a differential diagnosis using mutation detection of the ALPL gene for a variety of mutations [28].
4. Diagnosis
Laboratory assays
The laboratory assays for detecting different types of HPP primarily focus on measuring the activity of ALP and the concentrations of its substrates and products. These tests can help in diagnosing HPP, differentiating it from other disorders, and determining its type and severity. Key laboratory assays include:
ALP activity
The hallmark of HPP is low serum ALP activity, which is measured using routine biochemical assays. ALP activity is significantly lower than the normal range for the patient’s age and sex, aiding in the initial suspicion of HPP. In a recently published research, the prevalence of low AP activity and increased PLP levels was examined using 6,918,126 measurements collected between 2011 and 2016 from a single laboratory in northern Germany. The results revealed that 8.46% of the total measurements, displayed AP activity below 30 U/L. Within this subgroup, 6.09% exhibited elevated PLP levels. These results suggest that approximately 0.52% (1 in 194) of the subjects exhibited laboratory HPP. They proposed the automatic assessment of PLP levels in instances of low levels of ALP activity. Additionally, a notification to the prescribing physician advising the inclusion of HPP in the differential diagnosis and recommending further investigation was suggested [29].
Substrate accumulation tests
PEA: Elevated levels of PEA in urine indicate HPP. PEA is a natural substrate of ALP, and its accumulation in urine reflects ALP deficiency. A study specifically assessed the utility of urine PEA as a marker to diagnose and confirm HPP in adults and monitoring patients on ERT. The results of 59 patients, with and without confirmed HPP, were compared to other parameters. Urine PEA outperformed AP, suggesting it as a promising diagnostic and confirmatory marker for HPP [30].
PLP: Numerous studies have reported elevated levels of this substrate in HPP patients [7, 31]. In the absence of adequate ALP activity, PLP cannot be dephosphorylated to cross cell membranes effectively, leading to its accumulation. Therefore, high levels of PLP, the active form of vitamin B6, in the blood is another indicator of HPP. In addition, a correlation between the severity of the disease and the serum PLP level has been reported [32]. Assessing the levels of vitamin B6 metabolites, particularly in the context of seizures, can be useful. Elevated PLP levels in the context of seizures may prompt testing for HPP, especially in infants [16].
PPi: Increased concentrations of PPi in blood or urine can suggest HPP. PPi is an inhibitor of mineralization, and its accumulation contributes to the rickets or osteomalacia seen in HPP patients. As direct assays for PPi measurement are not currently available in clinical practice, understanding this correlation becomes crucial for assessing an individual’s fracture risk. This aligns with the observation that individuals with lower ALP activity tend to exhibit significantly higher lumbar spine BMD [33].
Calcium and phosphorus levels
While not diagnostic on their own, abnormal levels of calcium and phosphorus in the blood can indicate disruptions in bone metabolism associated with HPP. Hypercalcemia and hyperphosphatemia may occur, particularly in severe forms of HPP [34].
Bone specific AP (BSAP)
Measuring BSAP, a form of ALP expressed in bone, can provide additional insights into bone metabolism and the impact of HPP on bone formation [35].
Radiographic findings and BMD
While not laboratory assays, radiographic examinations, and BMD assessments can support the diagnosis of HPP by revealing characteristic skeletal abnormalities associated with the disease [33]. However, some studies reported that non-osteoporotic fractures may show higher than normal lumbar BMD recurrently in HPP patients and can be included as diagnostic criteria [36].
These assays, combined with clinical evaluation and radiographic findings, form the basis for diagnosing HPP and determining its severity and type. The diagnostic process must be comprehensive, considering both biochemical markers and clinical presentations.
Genetic testing
Confirmatory diagnosis of HPP requires genetic testing to identify mutations in the ALPL gene, which encodes the TNSALP. Various mutations can result in different forms of HPP, ranging from the perinatal lethal form to adult-onset forms. More than 400 mutations in the ALPL gene have been associated with HPP, adding complexity to genetic counseling. The intricacies arise from the dual modes of inheritance, incomplete penetrance in dominant forms, highly variable disease expression, even within families, and the presence of a benign prenatal form challenging to differentiate from severe cases [37]. Table 1 presents the overall presentations and mode of inheritance of each subtype of HPP.
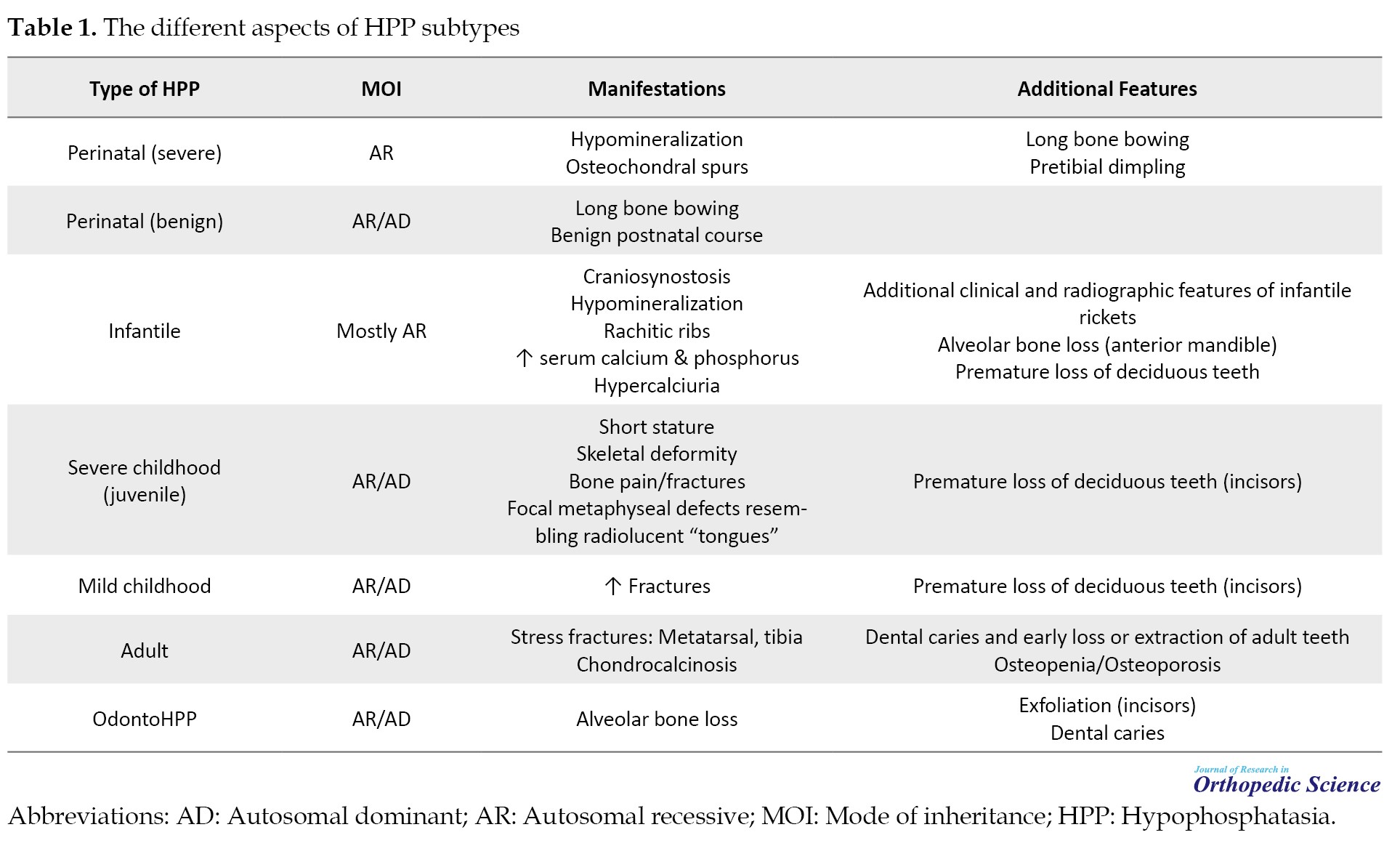
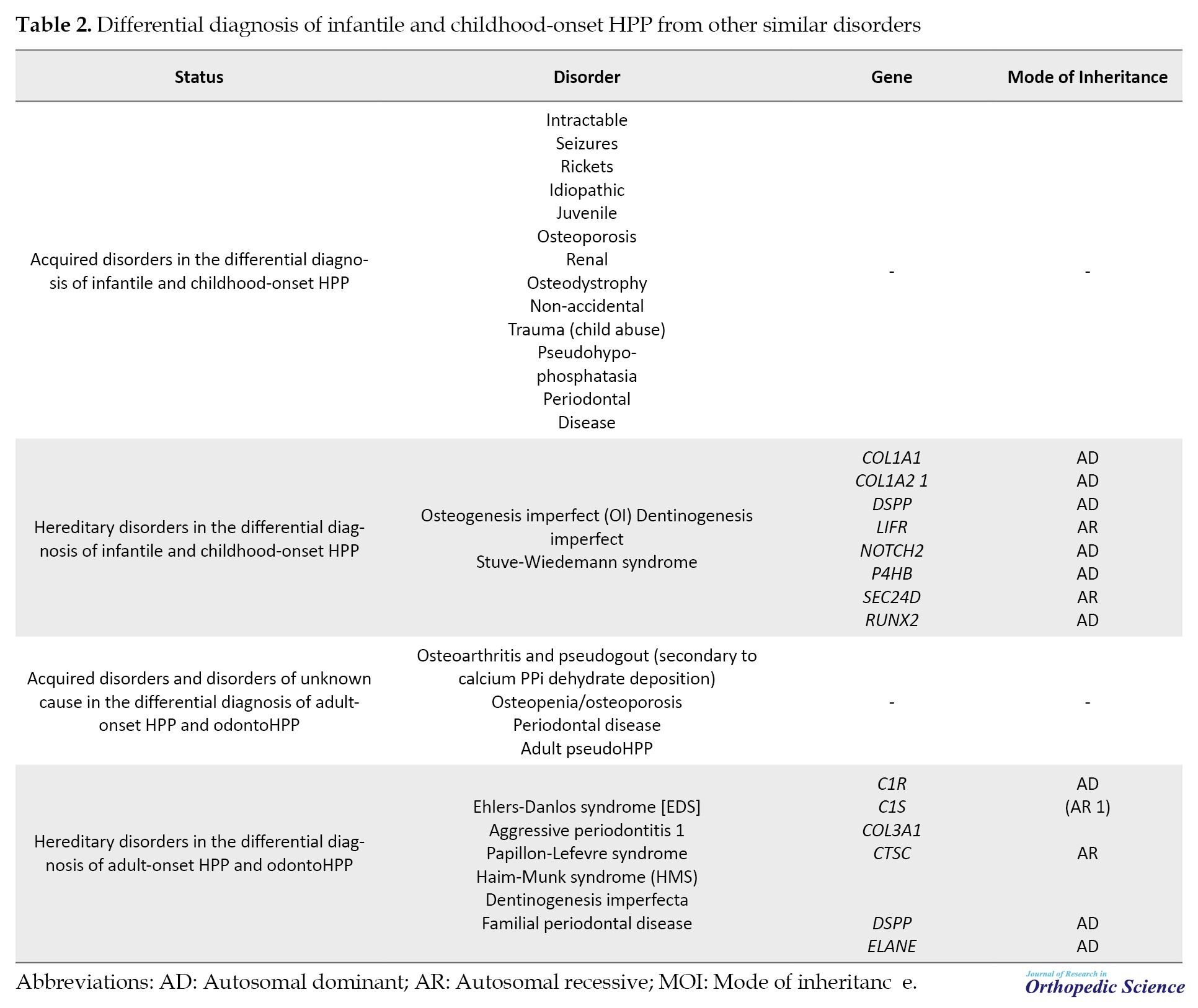
Loss-of-function mutations in the ALPL gene, responsible for encoding TNSALP, underlie the condition. The diverse array of missense mutations and the dominant negative impact of some mutations contribute significantly to clinical heterogeneity [38]. Comprehensive studies involving directed mutagenesis have enhanced our understanding of HPP’s cellular pathophysiology, aiding in classifying alleles based on severity and elucidating the dominant negative effect, offering molecular insights into dominant inheritance [6]. Genetic insights highlight distinct HPP categories: Rare, severe, and recessive HPP, and the more prevalent, likely underdiagnosed, mild recessive or mild dominant HPP. The prevalence of severe HPP is estimated at 1 in 300,000 in France and Northern Europe, while moderate forms may affect approximately 1 in 6 370 individuals [39]. Diagnosing HPP from other types of hereditary or acquired diseases with similar manifestations should be confirmed by genetic testing and mutation detection of ALPL gene. Table 2 demonstrates the list of complicating childhood and adult HPP diagnosis criteria from other diseases with similar clinical presentations.
5. Treatment
Asfotase alfa, also known as Strensiq, represents a groundbreaking therapy for severe manifestations of HPP, gaining FDA approval in 2015 [40]. This recombinant AP has become the primary treatment for infants, children, and select adults with severe HPP symptoms. The advent of targeted enzyme replacement therapy using asfotase alfa has marked a significant stride in the last decade, showcasing efficacy in restoring skeletal mineralization and achieving developmental milestones, ultimately improving mortality rates when compared to historical cohorts [41].
Contrarily, antiresorptive treatments are cautioned against in HPP patients due to their potential to lower serum AP levels, stemming from their inhibitory impact on osteoclasts and the possible predisposition to AFF. Indications for asfotase alfa treatment in adults involve a history of childhood involvement (before age 18 years) and specific criteria such as musculoskeletal pain requiring prescription medications, disabling polyarthropathy or chondrocalcinosis, major low-trauma fractures attributed to HPP, delayed fracture healing, orthopedic surgeries for HPP complications, functional impairment, low bone density, and nephrocalcinosis [13]. However, guidelines for patient selection and optimal therapy duration in adults with HPP are currently lacking. In a study involving 10 adult HPP patients, teriparatide was utilized to boost osteoblast production of AP, resulting in varied effects on bone density and reported fracture repair in some cases [42]. The application of the anti-sclerostin monoclonal antibody BPS804 has shown promise in enhancing BMD and regeneration in adult HPP individuals [43].
Regular dental monitoring is crucial for all individuals with HPP. Despite being primarily a physical ailment, HPP has been found to impact dental health in various studies. Several other treatments, including zinc, magnesium, cortisone, and plasma, have been explored with less encouraging therapeutic effects [44]. Bone marrow transplant effects in HPP appear transient, as bone lesions may reoccur around six months post-transplantation. Vitamin B6 supplementation has demonstrated improvement in neonatal seizures associated with HPP [45].
6. HPP and AFF
Studies indicate that AFF can occur in up to 10% of patients with HPP. This higher incidence emphasizes the importance of monitoring and managing bone health in individuals with HPP to minimize the risk of fractures [26]. Contrary to conventional approaches in treating bone-related disorders, antiresorptive medications, which impede bone breakdown, are explicitly contraindicated in HPP. The intricacies of HPP’s pathophysiology, notably the compromised function of TNSALP, render antiresorptive drugs detrimental, exacerbating the underlying metabolic disturbances. This contraindication stems from their potential to further perturb serum ALP levels, thereby intensifying the risk of AFF in HPP patients [46]. While acknowledging the significance of low serum ALP as a potential marker for AFF risk in HPP, ongoing investigations seek to unravel the precise correlation. This exploration is crucial in refining risk stratification strategies and tailoring interventions to the unique needs of HPP patients [47].
Addressing the formidable challenge of stabilizing fractures in HPP, particularly in severely affected individuals, demands a nuanced understanding of the disease’s impact on bone quality and healing dynamics. Unlike conventional fractures resulting from external trauma, HPP-related fractures often manifest as latent pseudo-fractures, predominantly in the diaphyseal area of long bones. Effective fracture management necessitates a departure from conventional extramedullary devices, such as plates, as these may pose a higher risk of complications. Appropriately sized intramedullary nails emerge as a preferred option, showcasing potential benefits in navigating the intricacies of fracture healing in HPP patients [48]. This insight underscores the importance of tailored approaches in fracture care, aligning with the distinct pathophysiological landscape of HPP.
7. Conclusion
HPP constitutes a rare genetic disorder with diverse clinical presentations. Characterized by six distinct forms categorized by age of onset and severity—perinatal, infantile, childhood (severe and mild), adult, and odonto—HPP manifests variably, presenting diagnostic challenges. The diagnosis, reliant on clinical, radiographic, and laboratory assessments, entails crucial laboratory criteria, including low serum ALP, elevated urinary PEA, and serum PLP levels. Confirming the diagnosis requires the identification of mutations in the TNAP gene. Genetic testing assumes a pivotal role in confirming HPP diagnoses, elucidating the underlying genetic mutations responsible for TNSALP dysfunction. This serves a dual purpose: Substantiating the diagnosis and unraveling the intricate interplay between genetic variants and clinical presentations. The primary therapeutic avenue for severe HPP manifestations is Asfotase alfa (Strensiq), serving as a first-line treatment in infants, children, and select adults. Notably, antiresorptive treatments are strictly contraindicated in HPP, as they may exacerbate the underlying pathophysiology and elevate the risk of AFF. This holistic perspective underscores the multidimensional challenges and intricacies of HPP diagnosis and management, necessitating a comprehensive understanding of its diverse clinical manifestations and therapeutic considerations.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Mahmoodreza Sarikhani and Mozhdeh Zabihiyeganeh; Review literature: Mahmoodreza Sarikhani and Roshanak Shams; The original draft preparation: Roshanak Shams and Mozhdeh Zabihiyeganeh; Review and editing: Amir Aminian and Mahmoodreza Sarikhani; Final approval: All authors.Supervision: Mozhdeh Zabihiyeganeh.
Conflict of interest
The authors declared no conflicts of interest.
References
In 1948, John C. Rathbun first identified a condition characterized by rickets, seizures, and diminished levels of alkaline phosphatase (AP) in a patient, a condition later known as hypophosphatasia (HPP) [1]. HPP emerges due to mutations affecting the gene for the tissue-nonspecific isoenzyme of AP (TNSALP), presenting a broad spectrum of systemic metabolic disturbances [2]. The AP-liver (ALPL) gene, responsible for HPP, is predominantly active in the bones, liver, kidneys, and developing teeth, still, its expression can also be found in the central nervous system, fibroblasts, endothelial cells, and several other cell types [3]. Despite its rarity, HPP is a significant yet often overlooked cause of osteomalacia and susceptibility to atypical fractures in adults, leading to a critical need for heightened awareness among healthcare professionals. This awareness is crucial to avoid the inappropriate use of antiresorptive therapies, such as bisphosphonates and denosumab, in these patients. Such treatments, intended for osteoporosis, can heighten the risk of atypical femoral fractures (AFF). The overarching goal of this comprehensive review is not only to enhance awareness and understanding among healthcare professionals, particularly in orthopedics and rheumatology but also to lay the groundwork for the development of evidence-based clinical practices. By consolidating recent advances and highlighting the necessity for appropriate therapeutic approaches, this study endeavors to improve patient outcomes and prevent the mismanagement of this complex condition.
2. Genetics and Molecular Pathophysiology
The molecular pathophysiology and pathogenesis of HPP are primarily centered on mutations in the ALPL gene (MIM*171760), which encodes the TNSALP, UniprotKB# P05186). This enzyme is crucial for bone and dental mineralization, and its dysfunction leads to the clinical manifestations of HPP. HPP results from mutations in the ALPL gene located on the short arm of chromosome 1 (1p36.1-34) [4]. These mutations can be heterozygous, compound heterozygous, or homozygous, with over 400 different mutations identified, including missense/nonsense mutations, deletions, and insertions [5]. A recent investigation analyzed the genetic inheritance patterns within a group of 424 individuals diagnosed with HPP. The results of this study indicated that the genetic characteristics of this cohort can be classified into three distinct clinical categories, severe HPP, which is both recessive and infrequent; moderate HPP, occurring more frequently and inheritable as either recessive or dominant; and mild HPP, the most common form, predominantly inherited in a dominant manner, marked by decreased levels of AP and nonspecific clinical symptoms [6]. However, mutations can lead to impaired function of the enzyme produced by TNSALP. This inborn metabolic disorder causes the extracellular buildup of natural substrates of TNSALP, including phosphoethanolamine (PEA), an integral part of the phosphatidylinositol-glycan linkage apparatus linking APs and other proteins to plasma membrane surfaces. Another substrate is inorganic pyrophosphate (PPi), recognized for hindering hydroxyapatite crystal formation and impacting biomineralization. Additionally, pyridoxal 5′-phosphate (PLP), the primary circulating form of vitamin B6 (B6), accumulates [7]. TNSALP is a glycosylphosphatidylinositol-anchored membrane enzyme expressed in various tissues, including bone, liver, kidneys, and teeth. It is involved in the dephosphorylation of several substrates, such as inorganic PPi, a natural inhibitor of hydroxyapatite crystal formation and growth [8]. PPi plays a pivotal role in regulating bone mineralization. It prevents excessive crystal growth, ensuring proper bone formation and remodeling. TNSALP’s activity is essential for maintaining the balance between PPi and phosphate (Pi) levels, facilitating normal bone mineralization [8]. Mutations in the ALPL gene reduce TNSALP activity, leading to the accumulation of PPi. High levels of PPi inhibit the deposition of calcium and Pi into the bone matrix, causing hypomineralization of bone and teeth, the hallmark of HPP. The mineralization defect results in a spectrum of skeletal abnormalities, ranging from severe perinatal forms with almost no bone mineralization to milder adult forms characterized by recurrent fractures, osteomalacia, and dental issues [9].
3. Clinical Presentations
HPP displays a diverse clinical spectrum, encompassing severe manifestations in infants, including life-threatening conditions and even fetal death, to milder forms observed in adults, characterized by dental or skeletal symptoms [10]. The classification of HPP involves six distinct forms, determined by factors, such as the age of onset, the severity of symptoms, and the specific clinical manifestations exhibited by individuals [3]. This classification system aids in understanding the varied presentations of HPP across different age groups and the severity of the condition.
Benign prenatal HPP
The prenatal benign variant of HPP is identifiable through gestational ultrasonography, revealing characteristics, such as shortened long bones and hypomineralization [1]. These manifestations often exhibit spontaneous improvement during late pregnancy or postnatal development. The natural progression of this form is unpredictable, with the potential for evolving from odonto HPP to the more severe infantile form [3]. The variability in its natural course underscores the challenge of foreseeing the trajectory of this prenatal benign form.
Perinatal (sever) HPP
This form, recognized as the most critical form, is primarily inherited through autosomal recessive traits and is considered rare. The clinical manifestations typically become evident by the end of pregnancy or at birth [3, 11], with a prognosis that is unfortunately almost always fatal shortly thereafter [10]. Radiological examinations reveal characteristic changes, including bone hypomineralization, radiolucent regions resembling “tongues” at the ends of long bones, slender and deformed bones, metaphyses presenting a “moth-eaten” and cup-shaped appearance, and the presence of osteochondral spurs [12]. Additional features encompass a high-pitched cry, pyridoxine-dependent seizures, periodic apnea accompanied by cyanosis and bradycardia, irritability, unexplained fever, myelophthisic anemia, and intracranial hemorrhage [10]. The serum ALP levels in neonates with this form are typically excessively low or undetectable [3]. Research results indicate that most mutations associated with this particular subtype are missense mutations. Establishing a correlation between genotype and phenotype can contribute to the development of a novel classification system. Such a classification of ALPL variants holds the potential to enhance the accuracy of distinguishing between perinatal lethal cases and other types of HPP [13, 14].
Infantile HPP
This form of HPP represents a moderately severe phenotype and is commonly associated with autosomal recessive inheritance [3]. Although newborns affected by this type may seem healthy at birth, clinical manifestations typically become apparent within the initial 6 months of life [15]. As mentioned, HPP causes the extracellular buildup of natural substrates of TNSALP, including PEA, PPi, and PLP. An established complication arises in life-threatening HPP during infancy due to compromised dephosphorylation of PLP to pyridoxal (PL) by TNSALP. PL, a B6 vitamer capable of crossing membranes, is then rephosphorylated to PLP or pyridoxamine Pi for intracellular enzymatic reactions. This compromised dephosphorylation process can result in B6-dependent seizures in infancy [16]. On the other hand, the increased excretion of calcium may contribute to renal damage. Despite the presence of an open fontanelle, premature craniosynostosis is frequently observed, potentially leading to elevated intracranial pressure [15]. Historically, it has been estimated that around 50% of infants with this subtype do not survive infancy [17]. A poor prognosis is often associated with the presence of respiratory failures, such as rib fractures, thoracic deformity, recurrent pneumonias, and or pyridoxine-dependent seizures [3].
Childhood HPP
Childhood HPP represents a highly heterogeneous clinical entity within the broader spectrum of HPP [15], exhibiting a notable diversity further categorized into mild and severe forms. In instances of mild presentation, affected children typically demonstrate good overall health, normal physical functionality, and minimal or negligible symptoms [18]. However, early-onset tooth loss is a characteristic feature, accompanied by subtle skeletal alterations, such as diminished bone mass evident in radiographic assessments. Conversely, severe childhood HPP, typically inherited as an autosomal recessive trait, manifests more prominently with challenges [19]. Patients experience premature tooth loss concomitant with skeletal pain, precipitating episodes of unexplained crying, and reluctance to ambulate. This muscular weakness contributes to delayed ambulation, a characteristic waddling gait, and difficulties in ascending stairs. Rachitic deformities may manifest as cranial misshaping, costochondral junction beading, bowed or knock-kneed legs, and joint enlargement due to metaphyseal flaring. While spontaneous remission of bone disease is acknowledged, the condition may reoccur during middle or late adulthood [20].
Adult HPP
The manifestation of HPP in adults emerges in middle age, with diagnoses occurring in individuals aged 18 years or older [15]. However, it is noteworthy that many adults with HPP have histories of signs or symptoms preceding this age that were not recognized during childhood, leading to significant delays in diagnosis [21]. Approximately 40%–55% of adults with HPP have a documented history of fractures, often multiple. At presentation, nearly two-thirds of patients exhibit symptoms, with musculoskeletal pain being a prevalent complaint (40%–75% in various series), affecting diverse areas, such as the feet, ankles, knees, thighs, hips, back, and joints [22]. Fractures commonly occur in the feet and femur/hip, but also in the wrists, vertebrae, or other bones, and are characterized by slow healing and potential recurrence. Bone mineral density (BMD) can vary, being low, normal, or high, with high lumbar spine BMD paradoxically associated with a higher risk of fractures. Dentition is also affected, with early loss of adult teeth being a common feature (25–35%) [2, 3]. Osteomalacia and pseudofractures, particularly in the femur, may precede HPP diagnosis, leading to bowing deformities of long bones in about 15% of patients. Radiographic calcific periarthritis, chondrocalcinosis, ossification of ligaments, scoliosis, and other symptoms, such as headaches, chronic fatigue, and gait abnormalities are observed in varying percentages [23]. Adult HPP, especially mild forms, may remain asymptomatic, contributing to underdiagnosis, as symptoms like musculoskeletal pain are non-specific and prevalent in the population. A study recently highlighted the clinical presentation and genetic profile of HPP in a cohort of 19 Chinese adults. The participants’ median age was 62, ranging from 32 to 74 years, with a predominance of female patients (16 out of 19). The most frequently reported issues were musculoskeletal symptoms (63.2%), dental complications (42.1%), fractures (36.8%), and fatigue (31.6%). Nearly half of the patients (47.4%) were incorrectly diagnosed with osteoporosis, and a third of them had undergone treatment with anti-resorptive drugs. The average serum ALP level among the group was notably low at 29.1 U/L, with 94.7% (18 out of 19) displaying ALP levels beneath the 40 U/L threshold. Genetic testing unveiled 14 different mutations in the ALPL gene, three of which were previously unreported [24]. Therefore, the diagnostic challenge arises in distinguishing between adult HPP and a normal phenotype, particularly in patients heterozygous for an ALPL gene mutation. The mentioned study has reported that the symptoms of two patients with compound heterozygous mutations were more severe than those with heterozygous mutations. Debilitating consequences of adult HPP include recurrent fractures, skeletal and joint pain, and muscle weakness. Notably, AFF can occur in up to 10% of HPP patients, with a higher frequency observed in those exposed to antiresorptive drugs before AFF onset [25, 26].
Odonto HPP
OdontoHPP is identified as the least severe yet most prevalent phenotype of HPP, affecting individuals across a broad age spectrum, encompassing both pediatric and adult populations [15]. This variant is primarily distinguished by its dental manifestations, which occur in the absence of radiographic or histopathological evidence indicative of rickets or osteomalacia. For instance, a recent study documented the case of a 2-year-old Japanese child diagnosed with odontoHPP. The child experienced premature loss of deciduous teeth, with the roots remaining intact. Serum analysis revealed low levels of AP alongside significantly elevated levels of PEA and PLP. Genetic testing of the ALPL gene identified a heterozygous mutation (NM_000478.6:c.1151C > A, p.Thr384Lys) [27]. Clinical presentations of these patients include the asymptomatic, premature loss of primary incisors, where the dental roots remain intact, and this occurs without accompanying gingival inflammation, ulceration, dental abscess formation, or a documented history of trauma. In the diagnostic assessment of patients presenting with early onset (before age 5 years) of unexplained tooth loss or the presence of abnormally loose teeth upon dental examination, odontoHPP should be considered as a differential diagnosis using mutation detection of the ALPL gene for a variety of mutations [28].
4. Diagnosis
Laboratory assays
The laboratory assays for detecting different types of HPP primarily focus on measuring the activity of ALP and the concentrations of its substrates and products. These tests can help in diagnosing HPP, differentiating it from other disorders, and determining its type and severity. Key laboratory assays include:
ALP activity
The hallmark of HPP is low serum ALP activity, which is measured using routine biochemical assays. ALP activity is significantly lower than the normal range for the patient’s age and sex, aiding in the initial suspicion of HPP. In a recently published research, the prevalence of low AP activity and increased PLP levels was examined using 6,918,126 measurements collected between 2011 and 2016 from a single laboratory in northern Germany. The results revealed that 8.46% of the total measurements, displayed AP activity below 30 U/L. Within this subgroup, 6.09% exhibited elevated PLP levels. These results suggest that approximately 0.52% (1 in 194) of the subjects exhibited laboratory HPP. They proposed the automatic assessment of PLP levels in instances of low levels of ALP activity. Additionally, a notification to the prescribing physician advising the inclusion of HPP in the differential diagnosis and recommending further investigation was suggested [29].
Substrate accumulation tests
PEA: Elevated levels of PEA in urine indicate HPP. PEA is a natural substrate of ALP, and its accumulation in urine reflects ALP deficiency. A study specifically assessed the utility of urine PEA as a marker to diagnose and confirm HPP in adults and monitoring patients on ERT. The results of 59 patients, with and without confirmed HPP, were compared to other parameters. Urine PEA outperformed AP, suggesting it as a promising diagnostic and confirmatory marker for HPP [30].
PLP: Numerous studies have reported elevated levels of this substrate in HPP patients [7, 31]. In the absence of adequate ALP activity, PLP cannot be dephosphorylated to cross cell membranes effectively, leading to its accumulation. Therefore, high levels of PLP, the active form of vitamin B6, in the blood is another indicator of HPP. In addition, a correlation between the severity of the disease and the serum PLP level has been reported [32]. Assessing the levels of vitamin B6 metabolites, particularly in the context of seizures, can be useful. Elevated PLP levels in the context of seizures may prompt testing for HPP, especially in infants [16].
PPi: Increased concentrations of PPi in blood or urine can suggest HPP. PPi is an inhibitor of mineralization, and its accumulation contributes to the rickets or osteomalacia seen in HPP patients. As direct assays for PPi measurement are not currently available in clinical practice, understanding this correlation becomes crucial for assessing an individual’s fracture risk. This aligns with the observation that individuals with lower ALP activity tend to exhibit significantly higher lumbar spine BMD [33].
Calcium and phosphorus levels
While not diagnostic on their own, abnormal levels of calcium and phosphorus in the blood can indicate disruptions in bone metabolism associated with HPP. Hypercalcemia and hyperphosphatemia may occur, particularly in severe forms of HPP [34].
Bone specific AP (BSAP)
Measuring BSAP, a form of ALP expressed in bone, can provide additional insights into bone metabolism and the impact of HPP on bone formation [35].
Radiographic findings and BMD
While not laboratory assays, radiographic examinations, and BMD assessments can support the diagnosis of HPP by revealing characteristic skeletal abnormalities associated with the disease [33]. However, some studies reported that non-osteoporotic fractures may show higher than normal lumbar BMD recurrently in HPP patients and can be included as diagnostic criteria [36].
These assays, combined with clinical evaluation and radiographic findings, form the basis for diagnosing HPP and determining its severity and type. The diagnostic process must be comprehensive, considering both biochemical markers and clinical presentations.
Genetic testing
Confirmatory diagnosis of HPP requires genetic testing to identify mutations in the ALPL gene, which encodes the TNSALP. Various mutations can result in different forms of HPP, ranging from the perinatal lethal form to adult-onset forms. More than 400 mutations in the ALPL gene have been associated with HPP, adding complexity to genetic counseling. The intricacies arise from the dual modes of inheritance, incomplete penetrance in dominant forms, highly variable disease expression, even within families, and the presence of a benign prenatal form challenging to differentiate from severe cases [37]. Table 1 presents the overall presentations and mode of inheritance of each subtype of HPP.
Loss-of-function mutations in the ALPL gene, responsible for encoding TNSALP, underlie the condition. The diverse array of missense mutations and the dominant negative impact of some mutations contribute significantly to clinical heterogeneity [38]. Comprehensive studies involving directed mutagenesis have enhanced our understanding of HPP’s cellular pathophysiology, aiding in classifying alleles based on severity and elucidating the dominant negative effect, offering molecular insights into dominant inheritance [6]. Genetic insights highlight distinct HPP categories: Rare, severe, and recessive HPP, and the more prevalent, likely underdiagnosed, mild recessive or mild dominant HPP. The prevalence of severe HPP is estimated at 1 in 300,000 in France and Northern Europe, while moderate forms may affect approximately 1 in 6 370 individuals [39]. Diagnosing HPP from other types of hereditary or acquired diseases with similar manifestations should be confirmed by genetic testing and mutation detection of ALPL gene. Table 2 demonstrates the list of complicating childhood and adult HPP diagnosis criteria from other diseases with similar clinical presentations.
5. Treatment
Asfotase alfa, also known as Strensiq, represents a groundbreaking therapy for severe manifestations of HPP, gaining FDA approval in 2015 [40]. This recombinant AP has become the primary treatment for infants, children, and select adults with severe HPP symptoms. The advent of targeted enzyme replacement therapy using asfotase alfa has marked a significant stride in the last decade, showcasing efficacy in restoring skeletal mineralization and achieving developmental milestones, ultimately improving mortality rates when compared to historical cohorts [41].
Contrarily, antiresorptive treatments are cautioned against in HPP patients due to their potential to lower serum AP levels, stemming from their inhibitory impact on osteoclasts and the possible predisposition to AFF. Indications for asfotase alfa treatment in adults involve a history of childhood involvement (before age 18 years) and specific criteria such as musculoskeletal pain requiring prescription medications, disabling polyarthropathy or chondrocalcinosis, major low-trauma fractures attributed to HPP, delayed fracture healing, orthopedic surgeries for HPP complications, functional impairment, low bone density, and nephrocalcinosis [13]. However, guidelines for patient selection and optimal therapy duration in adults with HPP are currently lacking. In a study involving 10 adult HPP patients, teriparatide was utilized to boost osteoblast production of AP, resulting in varied effects on bone density and reported fracture repair in some cases [42]. The application of the anti-sclerostin monoclonal antibody BPS804 has shown promise in enhancing BMD and regeneration in adult HPP individuals [43].
Regular dental monitoring is crucial for all individuals with HPP. Despite being primarily a physical ailment, HPP has been found to impact dental health in various studies. Several other treatments, including zinc, magnesium, cortisone, and plasma, have been explored with less encouraging therapeutic effects [44]. Bone marrow transplant effects in HPP appear transient, as bone lesions may reoccur around six months post-transplantation. Vitamin B6 supplementation has demonstrated improvement in neonatal seizures associated with HPP [45].
6. HPP and AFF
Studies indicate that AFF can occur in up to 10% of patients with HPP. This higher incidence emphasizes the importance of monitoring and managing bone health in individuals with HPP to minimize the risk of fractures [26]. Contrary to conventional approaches in treating bone-related disorders, antiresorptive medications, which impede bone breakdown, are explicitly contraindicated in HPP. The intricacies of HPP’s pathophysiology, notably the compromised function of TNSALP, render antiresorptive drugs detrimental, exacerbating the underlying metabolic disturbances. This contraindication stems from their potential to further perturb serum ALP levels, thereby intensifying the risk of AFF in HPP patients [46]. While acknowledging the significance of low serum ALP as a potential marker for AFF risk in HPP, ongoing investigations seek to unravel the precise correlation. This exploration is crucial in refining risk stratification strategies and tailoring interventions to the unique needs of HPP patients [47].
Addressing the formidable challenge of stabilizing fractures in HPP, particularly in severely affected individuals, demands a nuanced understanding of the disease’s impact on bone quality and healing dynamics. Unlike conventional fractures resulting from external trauma, HPP-related fractures often manifest as latent pseudo-fractures, predominantly in the diaphyseal area of long bones. Effective fracture management necessitates a departure from conventional extramedullary devices, such as plates, as these may pose a higher risk of complications. Appropriately sized intramedullary nails emerge as a preferred option, showcasing potential benefits in navigating the intricacies of fracture healing in HPP patients [48]. This insight underscores the importance of tailored approaches in fracture care, aligning with the distinct pathophysiological landscape of HPP.
7. Conclusion
HPP constitutes a rare genetic disorder with diverse clinical presentations. Characterized by six distinct forms categorized by age of onset and severity—perinatal, infantile, childhood (severe and mild), adult, and odonto—HPP manifests variably, presenting diagnostic challenges. The diagnosis, reliant on clinical, radiographic, and laboratory assessments, entails crucial laboratory criteria, including low serum ALP, elevated urinary PEA, and serum PLP levels. Confirming the diagnosis requires the identification of mutations in the TNAP gene. Genetic testing assumes a pivotal role in confirming HPP diagnoses, elucidating the underlying genetic mutations responsible for TNSALP dysfunction. This serves a dual purpose: Substantiating the diagnosis and unraveling the intricate interplay between genetic variants and clinical presentations. The primary therapeutic avenue for severe HPP manifestations is Asfotase alfa (Strensiq), serving as a first-line treatment in infants, children, and select adults. Notably, antiresorptive treatments are strictly contraindicated in HPP, as they may exacerbate the underlying pathophysiology and elevate the risk of AFF. This holistic perspective underscores the multidimensional challenges and intricacies of HPP diagnosis and management, necessitating a comprehensive understanding of its diverse clinical manifestations and therapeutic considerations.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization: Mahmoodreza Sarikhani and Mozhdeh Zabihiyeganeh; Review literature: Mahmoodreza Sarikhani and Roshanak Shams; The original draft preparation: Roshanak Shams and Mozhdeh Zabihiyeganeh; Review and editing: Amir Aminian and Mahmoodreza Sarikhani; Final approval: All authors.Supervision: Mozhdeh Zabihiyeganeh.
Conflict of interest
The authors declared no conflicts of interest.
References
- Reis FS, Lazaretti-Castro M. Hypophosphatasia: From birth to adulthood. Arch Endocrinol Metab. 2023; 67(5):e000626. [DOI:10.20945/2359-3997000000626] [PMID] [PMCID]
- Shapiro JR, Lewiecki EM. Hypophosphatasia in adults: clinical assessment and treatment considerations. J Bone Miner Res. 2017; 32(10):1977-80. [DOI:10.1002/jbmr.3226] [PMID]
- Tournis S, Yavropoulou MP, Polyzos SA, Doulgeraki A. Hypophosphatasia. J Clin Med. 2021; 10(23):5676. [DOI:10.3390/jcm10235676] [PMID] [PMCID]
- Greenberg CR, Evans JA, McKendry-Smith S, Redekopp S, Haworth JC, Mulivor R, et al. Infantile hypophosphatasia: Localization within chromosome region 1p36. 1-34 and prenatal diagnosis using linked DNA markers. Am J Hum Genet. 1990; 46(2):286-92. [PMID]
- Villa-Suárez JM, García-Fontana C, Andújar-Vera F, González-Salvatierra S, de Haro-Muñoz T, Contreras-Bolívar V, et al. Hypophosphatasia: A unique disorder of bone mineralization. Int J Mol Sci. 2021; 22(9):4303. [DOI:10.3390/ijms22094303] [PMID] [PMCID]
- Mornet E, Taillandier A, Domingues C, Dufour A, Benaloun E, Lavaud N, et al. Hypophosphatasia: A genetic-based nosology and new insights in genotype-phenotype correlation. Eur J Hum Genet. 2021; 29(2):289-99. [DOI:10.1038/s41431-020-00732-6] [PMID] [PMCID]
- Whyte MP, Zhang F, Wenkert D, Mack KE, Bijanki VN, Ericson KL, et al. Hypophosphatasia: Vitamin B6 status of affected children and adults. Bone. 2022; 154:116204. [DOI:10.1016/j.bone.2021.116204] [PMID]
- Bangura A, Wright L, Shuler T. Hypophosphatasia: current literature for pathophysiology, clinical manifestations, diagnosis, and treatment. Cureus. 2020; 12(6):e8594. [DOI:10.7759/cureus.8594] [PMID] [PMCID]
- Salles JP. Hypophosphatasia: Biological and clinical aspects, avenues for therapy. Clin Biochem Rev. 2020; 41(1):13-27.[DOI:10.33176/AACB-19-00031] [PMID] [PMCID]
- Simon S, Resch H, Klaushofer K, Roschger P, Zwerina J, Kocijan R. Hypophosphatasia: From diagnosis to treatment. Curr Rheumatol Rep. 2018; 20(11):69. [DOI:10.1007/s11926-018-0778-5] [PMID]
- Duffus S, Thrasher B, Calikoglu AS. Brief clinical report: Hypophosphatasia-diagnostic considerations and treatment outcomes in an infant. Case Rep Pediatr. 2018; 2018:5719761.[DOI:10.1155/2018/5719761] [PMID] [PMCID]
- Offiah AC, Vockley J, Munns CF, Murotsuki J. Differential diagnosis of perinatal hypophosphatasia: Radiologic perspectives. Pediatr Radiol. 2019; 49(1):3-22. [DOI:10.1007/s00247-018-4239-0] [PMID] [PMCID]
- Whyte MP. Hypophosphatasia: An overview For 2017. Bone. 2017; 102:15-25. [DOI:10.1016/j.bone.2017.02.011] [PMID]
- Sperelakis-Beedham B, Taillandier A, Domingues C, Guberto M, Colin E, Porquet-Bordes V, et al. Utility of genetic testing for prenatal presentations of hypophosphatasia. Mol Genet Metab. 2021; 132(3):198-203. [DOI:10.1016/j.ymgme.2021.01.009] [PMID]
- Mornet E. Hypophosphatasia. Best Pract Res Clin Rheumatol. 2008; 22(1):113-27. [DOI:10.1016/j.berh.2007.11.003] [PMID]
- Whyte MP, May JD, McAlister WH, Burgener K, Cortez SR, Kreienkamp R, et al. Vitamin B6 deficiency with normal plasma levels of pyridoxal 5′-phosphate in perinatal hypophosphatasia. Bone. 2021; 150:116007. [DOI:10.1016/j.bone.2021.116007] [PMID]
- Vogt M, Girschick H, Schweitzer T, Benoit C, Holl-Wieden A, Seefried L, et al. Pediatric hypophosphatasia: Lessons learned from a retrospective single-center chart review of 50 children. Orphanet J Rare Dis. 2020; 15(1):212. [DOI:10.1186/s13023-020-01500-x] [PMID] [PMCID]
- Silva I, Castelão W, Mateus M, Branco J. Childhood hypophosphatasia with myopathy: Clinical report with recent update. Acta Reumatol Port. 2012; 37(1):92-6. [PMID]
- Moulin P, Vaysse F, Bieth E, Mornet E, Gennero I, Dalicieux-Laurencin S, et al. Hypophosphatasia may lead to bone fragility: Don’t miss it. Eur J Pediatr. 2009; 168(7):783-8. [DOI:10.1007/s00431-008-0835-6] [PMID]
- Rush ET. Childhood hypophosphatasia: To treat or not to treat. Orphanet J Rare Dis. 2018; 13(1):116. [DOI:10.1186/s13023-018-0866-7] [PMID] [PMCID]
- Högler W, Langman C, Gomes da Silva H, Fang S, Linglart A, Ozono K, et al. Diagnostic delay is common among patients with hypophosphatasia: Initial findings from a longitudinal, prospective, global registry. BMC Musculoskelet Disord. 2019; 20(1):80. [DOI:10.1186/s12891-019-2420-8] [PMID] [PMCID]
- Seefried L, Dahir K, Petryk A, Högler W, Linglart A, Martos-Moreno GÁ, et al. Burden of illness in adults with hypophosphatasia: Data from the global hypophosphatasia patient registry. J Bone Miner Res. 2020; 35(11):2171-8.[DOI:10.1002/jbmr.4130] [PMID]
- Brandi ML, Khan AA, Rush ET, Ali DS, Al-Alwani H, Almonaei K, et al. The challenge of hypophosphatasia diagnosis in adults: Results from the HPP International Working Group Literature Surveillance. Osteoporos Int. 2024; 35(3):439-49. [PMID]
- Li X, Ren N, Wang Z, Wang Y, Hu Y, Hu W, et al. Clinical and genetic characteristics of hypophosphatasia in Chinese adults. Genes. 2023; 14(4):922. [DOI:10.3390/genes14040922] [PMID] [PMCID]
- Tsiantouli E, Biver E, Chevalley T, Petrovic R, Hannouche D, Ferrari S. Prevalence of low serum alkaline phosphatase and hypophosphatasia in adult patients with atypical femur fractures. Calcif Tissue Int. 2022; 110(6):703-11.[DOI:10.1007/s00223-022-00949-1] [PMID] [PMCID]
- Charoenngam N, Thongpiya J, Yingchoncharoen P, Ponvilawan B, Marangoz MS, Chenbhanich J, et al. Atypical femoral fracture in hypophosphatasia: A systematic review. Int J Endocrinol. 2023; 2023:5544148. [DOI:10.1155/2023/5544148] [PMID] [PMCID]
- Oto Y, Suzuki D, Morita T, Inoue T, Nitta A, Murakami N, et al. A case report of odonto-hypophosphatasia with a novel variant in the ALPL gene. J Pediatr Endocrinol Metab. 2024; 37(3):276-9. [DOI:10.1515/jpem-2023-0549] [PMID]
- Jiang J, Li H, Kong H, Zeng X, Gou L, Xu J. Odontohypophosphatasia caused by a novel combination of two heterozygous variants: A case report. J Clin Pediatr Dent. 2023; 47(4):111-5. [PMID]
- Schmidt T, Schmidt C, Amling M, Kramer J, Barvencik F. Prevalence of low alkaline phosphatase activity in laboratory assessment: Is hypophosphatasia an underdiagnosed disease? Orphanet Journal of Rare Diseases. 2021; 16(1):452. [DOI:10.1186/s13023-021-02084-w] [PMID] [PMCID]
- Shajani-Yi Z, Ayala-Lopez N, Black M, Dahir KM. Urine phosphoethanolamine is a specific biomarker for hypophosphatasia in adults. Bone. 2022; 163:116504. [DOI:10.1016/j.bone.2022.116504] [PMID]
- Whyte MP, Zhang F, Mack KE, Wenkert D, Gottesman GS, Ericson KL, et al. Pyridoxine challenge reflects pediatric hypophosphatasia severity and thereby examines tissue-nonspecific alkaline phosphatase’s role in vitamin B6 metabolism. Bone. 2024; 181:117033. [DOI:10.1016/j.bone.2024.117033] [PMID]
- Mornet E. Hypophosphatasia. Metabolism. 2018; 82:142-55. [DOI:10.1016/j.metabol.2017.08.013] [PMID]
- Genest F, Claußen L, Rak D, Seefried L. Bone mineral density and fracture risk in adult patients with hypophosphatasia. Osteoporos Int. 2021; 32(2):377-85. [DOI:10.1007/s00198-020-05612-9] [PMID] [PMCID]
- Michigami T, Ohata Y, Fujiwara M, Mochizuki H, Adachi M, Kitaoka T, et al. Clinical practice guidelines for hypophosphatasia. Clin Pediatr Endocrinol. 2020; 29(1):9-24. [DOI:10.1297/cpe.29.9] [PMID] [PMCID]
- Riancho JA. Diagnostic approach to patients with low serum alkaline phosphatase. Calcif Tissue Int. 2023; 112(3):289-96. [DOI:10.1007/s00223-022-01039-y] [PMID]
- Sadhukhan S, Mehta P, Rajender S, Gupta SK, Chattopadhyay N. Proposing a clinical algorithm for better diagnosis of hypophosphatasia in resource-limiting situations.Osteoporos Int. 2022; 33(12):2479-93. [DOI:10.1007/s00198-022-06480-1] [PMID]
- Mornet E. Genetics of hypophosphatasia. Arch Pediatr. 2017; 24(5S2):5S51-6. [DOI:10.1016/S0929-693X(18)30014-9] [PMID]
- Liu M, Liu M, Liang X, Wu D, Li W, Su C, et al. Clinical and genetic characteristics of hypophosphatasia in Chinese children. Orphanet J Rare Dis. 2021; 16(1):159. [DOI:10.1186/s13023-021-01798-1] [PMID] [PMCID]
- Fenn JS, Lorde N, Ward JM, Borovickova I. Hypophosphatasia. J Clin Pathol. 2021; 74(10):635-40. [DOI:10.1136/jclinpath-2021-207426] [PMID]
- Whyte MP, Rockman-Greenberg C, Ozono K, Riese R, Moseley S, Melian A, et al. Asfotase Alfa treatment improves survival for perinatal and infantile hypophosphatasia. The J Clin Endocrinol Metab. 2016; 101(1):334-42. [DOI:10.1210/jc.2015-3462] [PMID] [PMCID]
- Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012; 366(10):904-13. [DOI:10.1056/NEJMoa1106173] [PMID]
- Schalin-Jäntti C, Mornet E, Lamminen A, Välimäki MJ. Parathyroid hormone treatment improves pain and fracture healing in adult hypophosphatasia. J Clin Endocrinol Metab. 2010; 95(12):5174-9. [DOI:10.1210/jc.2010-1168] [PMID]
- Seefried L, Baumann J, Hemsley S, Hofmann C, Kunstmann E, Kiese B, et al. Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia. J Clin Invest. 2017; 127(6):2148-58. [DOI:10.1172/JCI83731] [PMID] [PMCID]
- Weber TJ, Sawyer EK, Moseley S, Odrljin T, Kishnani PS. Burden of disease in adult patients with hypophosphatasia: Results from two patient-reported surveys. Metabolism. 2016; 65(10):1522-30. [DOI:10.1016/j.metabol.2016.07.006] [PMID]
- Plecko B, Stöckler S. Vitamin B6 dependent seizures. Can J Neurol Sci. 2009; 36 (Suppl 2):S73-7. [PMID]
- Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016; 12(4):233-46. [DOI:10.1038/nrendo.2016.14] [PMID]
- Bhattacharyya T, Jha S, Wang H, Kastner DL, Remmers EF. Hypophosphatasia and the risk of atypical femur fractures: A case-control study. BMC Musculoskelet Disord. 2016; 17:332. [DOI:10.1186/s12891-016-1191-8] [PMID] [PMCID]
- Genest F, Seefried L. Subtrochanteric and diaphyseal femoral fractures in hypophosphatasia-not atypical at all. Osteoporos Int. 2018; 29(8):1815-25. [DOI:10.1007/s00198-018-4552-3] [PMID]
Type of Study: Review Paper |
Subject:
Rheumatology
Received: 2022/02/25 | Accepted: 2022/03/25 | Published: 2022/11/1
Received: 2022/02/25 | Accepted: 2022/03/25 | Published: 2022/11/1
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |

Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
