

Volume 11, Issue 2 (May 2024)
JROS 2024, 11(2): 55-76 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Bagherifard A, Mokhtari K, Yahyazedeh H. Molecular Mechanisms and Bone Dynamics in Osteomyelitis: A Mini-review. JROS 2024; 11 (2) :55-76
URL: http://jros.iums.ac.ir/article-1-2267-en.html
URL: http://jros.iums.ac.ir/article-1-2267-en.html



1- Bone and Joint Reconstruction Research Center, Shafa Orthopedic Hospital, Iran University of Medical Sciences, Tehran, Iran.
2- Department of Cell and Molecular Biology and Microbiology, Faculty of Biological Science and Technology, University of Isfahan, Isfahan, Iran.
3- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Iran University of Medical Science, Tehran, Iran. & Department of Orthopedic Surgery, Faculty of Medicine, Farhikhtegan Hospital, Tehran Medical Sciences Branch, Islamic Azad University, Tehran, Iran.
2- Department of Cell and Molecular Biology and Microbiology, Faculty of Biological Science and Technology, University of Isfahan, Isfahan, Iran.
3- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Iran University of Medical Science, Tehran, Iran. & Department of Orthopedic Surgery, Faculty of Medicine, Farhikhtegan Hospital, Tehran Medical Sciences Branch, Islamic Azad University, Tehran, Iran.
Keywords: Osteomyelitis (OM), Molecular mechanisms, Bone dynamics, Osteoclasts, Osteoblasts, Inflammation, Bone resorption, Cytokines, Bone repair, Staphylococcus aureus
Full-Text [PDF 859 kb]
(347 Downloads)
| Abstract (HTML) (865 Views)
Full-Text: (339 Views)
Introduction
Bone
Bone is a highly dynamic tissue that continuously remodels itself to preserve homeostasis. This process is regulated by two distinct types of cells: Osteoblasts, which arise from mesenchymal stem cells and are responsible for forming new bone, and osteoclasts, which originate from hematopoietic precursors and mediate bone resorption. The differentiation of osteoclasts from monocytes is regulated by two cytokines: M-CSF and receptor activator of NF-κB ligand (RANKL). When these cytokines bind to their respective receptors on monocytes, they initiate the differentiation process into osteoclasts [1-3]. In the body, ongoing bone remodeling helps repair microfractures and compensates for wear caused by biomechanical stress, typically resulting in complete renewal within 7 to 10 months [4-6]. The skeletal system is highly sensitive to both local and systemic signals. Several diseases are characterized by heightened bone resorption, which may be associated with hormonal imbalances and aging in certain cases [7, 8].
Staphylococcus aureus
S. aureus is the leading pathogen responsible for osteomyelitis (OM) and is one of the most common microorganisms implicated in persistent infections [9, 10]. S. aureus interferes with immune cell functions and disrupts the equilibrium between osteoclasts and osteoblasts, leading to sustained infection and continuous bone degradation, which are critical factors in the progression of chronic OM [9]. S. aureus promotes chronic infection and bone destruction through several key mechanisms. It secretes virulence factors that enhance bacterial colonization, induce the death of immune cells, and alter the immune environment, allowing the bacteria to evade the immune system. These virulence factors are also associated with the formation of abscesses in OM. Additionally, S. aureus forms biofilms in response to vascular damage and reduced oxygen levels, creating a protective barrier that prevents immune cells from contacting the pathogen, impairs neutrophil activation, and encourages macrophage differentiation. The pathogen also adjusts its metabolism during infection, enabling it to compete with immune cells for energy and survive. Finally, S. aureus can survive within immune cells by escaping the phagosome and continuing to replicate within the cell [10-16].
Molecular insight into S. aureus
S. aureus infiltrates the cortical bone, infecting osteocytes. In response, these infected osteocytes release elevated levels of chemokines, including CCL5, CXCL1, CXCL8, CXCL9, and CXCL11. Neutrophils and T lymphocytes are activated by these chemokines, which in turn drive inflammation and contribute to the destruction of bone tissue. This immune response, while aimed at controlling the infection, also exacerbates bone damage, leading to the progression of OM [17]. S. aureus can also survive within osteoblasts and cause morphological changes or even cell death. Infected osteoblasts release higher levels of inflammatory factors, which promote the migration of immune cells and stimulate osteoclastogenesis [9, 10, 18].
Neutrophils
At the early stage of infection, neutrophils are the first immune cells to become activated and carry out their defensive functions at the site of infection [19]. However, during S. aureus-induced OM, key neutrophil functions—including chemotactic migration, activation, and phagocytic killing—are disrupted. This condition hinders the formation of neutrophil extracellular traps (NETs) and diminishes their ability to eliminate bacteria. Moreover, neutrophils undergo modifications during the infection that contribute to tissue damage and facilitate bacterial survival and spread at the infection site. Initially, host cells release higher levels of chemokines to attract neutrophils to the infection site, where they perform bactericidal functions. However, S. aureus secretes virulence factors such as chronic inflammatory proteins (CHIPs) and the FPRL1 inhibitory protein (FLIPr), which interfere with neutrophil chemotaxis in OM. CHIPs reduce the production of chemokines and impair neutrophil migration toward the site of infection, thereby hindering the host’s ability to combat bacterial invasion effectively [20, 21].
Neutrophil activation normally depends on the recognition of pathogen-specific receptors. However, Ssl3—a virulence factor secreted by S. aureus and part of the Ssl protein family—interferes with this process by inhibiting pathogen recognition. SSL3 inhibits neutrophil activation by competitively binding to TLR2, thus impairing an effective immune response. Additionally, the virulence factor CHIPs competes with complement C5a for binding, preventing C5a from activating neutrophils via its receptor [11, 22, 23]. In response to immune challenges, neutrophils release inhibitory proteins, such as neutrophil elastase, proteinase-3, and cathepsin G, to degrade CHIPs. However, S. aureus counteracts this defense mechanism by producing extracellular adherence protein (Eap) and its homologs, EapH1 and EapH2. These proteins inhibit the activity of neutrophil elastases, effectively protecting the bacteria from immune clearance and allowing them to persist in the infection site [24]. Moreover, in chronic infections, some neutrophils express MHC class II molecules, allowing them to present antigens to T cells. Research has shown that under conditions of persistent infection, these neutrophils can participate in antigen presentation and activate T cells, thus playing a role in the adaptive immune response [25].
A hallmark characteristic of S. aureus in OM is its ability to form biofilms on implant surfaces. When biofilms are present, the bacteria are protected from immune surveillance, unlike planktonic bacteria, which are rapidly detected and cleared by immune cells in the absence of biofilm formation. Biofilm formation enables bacteria to evade immune responses and persist at the site of infection. However, when planktonic bacteria adhere to implant surfaces, such as internal fixation devices (e.g. plates, screws), biofilms are formed, creating a protective niche that shields the bacteria from immune detection and promotes their persistence [13]. Immature biofilms do not provide a strong barrier and can still be penetrated by fully activated neutrophils, enabling them to perform their immune functions. However, as the biofilm matures, its ability to act as a physical shield against immune cells increases significantly. Extracellular polymeric substances (EPS), a vital component of biofilms, play a pivotal role in immunosuppression. EPS hinders neutrophil recognition of bacteria by redirecting their attention from the pathogen to the biofilm itself, thereby aiding the pathogen in evading the host’s immune system and promoting its persistence at the infection site [26, 27].
The ability of S. aureus to persistently survive within immune cells is a critical factor that impedes neutrophils from effectively performing phagocytosis and eliminating pathogens. S. aureus disrupts the function of neutrophil phagosomes, facilitating its survival within these immune cells [28-30]. In OM, S. aureus likely survives within the phagosome by disrupting the expression of LC3-modified proteins. Understanding the mechanisms by which S. aureus regulates LC3 expression in this context is essential. Targeting and inhibiting these pathways could offer a novel approach to restore phagosome function and enhance the ability of immune cells to eliminate pathogens efficiently [31].
The primary function of NETs is to capture and eliminate S. aureus at the site of infection. However, this process may result in damage to endothelial cells and other adjacent tissues, potentially contributing to additional tissue injury and complications [32]. Research has demonstrated that histone proteins mediate platelet activation through TLR4 and TLR2, thereby promoting blood coagulation and thrombin formation, a process linked to platelet-rich microthrombosis in sepsis models. Thus, histones found in NETs may play a pivotal role in the development of microthrombosis in chronic OM. This microthrombosis could impede tissue repair and create an environment conducive to the growth of S. aureus. Consequently, NETs not only serve to capture pathogens but also substantially contribute to tissue damage in chronic OM [33].
Macrophages
During the initial phase of infection, circulating monocytes differentiate into macrophages in response to the influence of inflammatory cytokines. M1 macrophages play a key role in pathogen elimination by releasing lysosomal enzymes and enhancing inflammation through the secretion of pro-inflammatory cytokines. In contrast, M2 macrophages secrete anti-inflammatory cytokines to modulate the inflammatory response and produce growth factors, such as platelet-derived growth factor and fibroblast growth factor, which contribute to tissue repair and healing [34]. In chronic OM, there is an imbalance and dysregulation in the interconversion and proportion of M1 and M2 macrophages. The prolonged presence of M1 macrophages contributes to an exaggerated inflammatory response, which can potentially cause significant tissue damage. On the other hand, an overproduction of M2 macrophages can impair effective phagocytosis and cytotoxic activity, thereby favoring biofilm formation and enhancing bacterial resistance, which hinders the resolution of infection [35].
These findings suggest that the STAT3/STAT6 pathway may impede the pathogen-clearing function of macrophages, thereby facilitating the persistence of OM. Additionally, IL-10 has been shown to activate the STAT3 pathway, which further suppresses the macrophage immune response. As a result, targeting this signaling pathway could offer a potential immunoregulatory strategy for treating chronic OM [36-38]. The biofilm formed by S. aureus also plays a crucial role in modulating macrophage polarization [39, 40].
In chronic OM, the migration of macrophages is regulated by the NF-κB/TWIST1 signaling pathway. This pathway plays a crucial role in modulating the movement and activation of macrophages, which are essential for the inflammatory response and tissue remodeling during the progression of the infection [35]. Additionally, TWIST1 enhances the expression of matrix metalloproteinases (MMP9 and MMP3), which facilitate macrophage migration by breaking down extracellular matrix components at the infection site. This degradation allows macrophages to move more effectively towards areas of infection, thereby contributing to the inflammatory response and tissue remodeling seen in chronic OM [41].
The PI3K/Akt-Beclin signaling pathway plays a crucial role in regulating macrophage autophagy, pathogen phagocytosis, and the NF-κB-mediated inflammatory response in S. aureus-induced OM. Recent studies have shown that inhibiting PI3K impairs macrophage autophagy, thereby reducing their ability to phagocytose S. aureus [42] effectively.
T cells
Upon encountering an antigen, T lymphocytes differentiate into two main subsets: Helper T cells (CD4+ T cells) and cytotoxic T cells (CD8+ T cells). CD4+ T cells bolster immune responses by releasing a variety of inflammatory mediators that activate other immune cells and regulate inflammation by controlling the proliferation of certain immune cell types. In contrast, CD8+ T cells target and eliminate pathogens directly by secreting cytotoxic proteins and cytokines, such as perforin and granzyme [26]. Dendritic cells play a crucial role in activating T cells through the presentation of antigens. However, leukocidin AB (LukAB), a virulence factor secreted by S. aureus, disrupts this process by inducing the death of dendritic cells, which are crucial for effective antigen presentation and the subsequent initiation of immune responses. This interference by LukAB undermines the immune system’s ability to recognize and respond to the pathogen [43] properly. CTLA-4, an inhibitory molecule predominantly expressed on T cells, functions by binding to CD80/CD86 molecules present on antigen-presenting cells (APCs). This binding prevents the interaction between CD28 on T cells and these costimulatory molecules, thereby inhibiting the activation of T cells. As a result, the immune response is compromised, impeding the clearance of pathogens and allowing the infection to persist [26, 44]. As OM transitions from the acute to the chronic phase, the functionality of CD8+ T cells is significantly impaired, causing them to contribute more to the pathogenesis of chronic OM rather than assisting in the elimination of pathogens. This dysfunction in CD8+ T cell activity enables the infection to persist and worsen over time [10].
Studies have demonstrated that in chronic OM, the cytotoxic function of CD8+ T cells is notably compromised due to prolonged exposure to elevated antigen levels and inflammatory mediators. These T cells, which exhibit immunosuppressive characteristics, are classified as exhausted CD8+ T cells [44]. Exhausted CD8+ T cells show a significant decline in the production of crucial cytokines, such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-12, IL-18, and IL-10. Additionally, the synthesis of cytotoxic proteins, including granzyme and perforin, is impaired, thereby compromising their ability to target and eliminate pathogens efficiently [45]. Importantly, exhausted CD8+ T cells express various inhibitory receptors, including PD-1, LAG-3, 2B4, CD160, TIM-3, and TIGHT, which contribute to their functional impairment and inability to combat pathogens [45-47] effectively. During chronic infections, inhibitory receptors continue to exert immunosuppressive effects. Although the impact of each receptor individually may be modest, exhausted T cells typically co-express several receptors, such as PD-1 and LAG-3. This co-expression leads to a synergistic effect, resulting in a more substantial immunosuppressive response than any single receptor could induce on its own [48]. Moreover, the metabolic profile of exhausted CD8+ T cells changes to adapt to the environment of persistent infection. These alterations enable the cells to survive and function under chronic stress conditions, though they may impair their full effector functions and contribute to immune dysfunction [49-53]. Recent studies have highlighted a notable distinction in the expression of transcription factors between normal effector CD8+ T cells and their exhausted counterparts. Transcription factors such as T-bet, EOMES, and TCF1 have gained recognition for their pivotal role in driving the functional shift of CD8+ T cells toward exhaustion. These factors are integral to regulating CD8+T cell functionality and facilitate their transition into an exhausted state, particularly in the context of chronic infections [10].
T-bet and EOMES are essential transcription factors that regulate the differentiation of naive CD8+ T cells. When T-bet expression surpasses that of EOMES, CD8+ T cells are directed towards differentiating into terminal effector cells, which are critical for executing immune responses. In contrast, when EOMES expression surpasses that of T-bet, naive CD8+ T cells are more likely to differentiate into memory T cells. However, sustained high expression of EOMES is associated with the progressive exhaustion of CD8+ T cells, leading to diminished immune function in chronic infections [54]. Some researchers propose that the sustained high expression of EOMES may be linked to elevated levels of PD-1, a marker known to be upregulated during chronic OM. This outcome suggests that the overexpression of EOMES may contribute to the exhaustion of CD8+ T cells by promoting PD-1 expression, which in turn impairs T cell function and supports the persistence of the infection [48, 55].
TCF1, a transcription factor exclusive to T cells, plays a pivotal role in the differentiation and sustained function of exhausted CD8+ T cells. During the initial stages of chronic infection, TCF1 is co-expressed with CXCL5 in CD8+ T cells. However, as the infection progresses, these cells progressively adopt exhausted characteristics, leading to a decline in their effector functions and contributing to the persistence of the infection. When TCF1 is knocked down in CD8+ T cells, there is a tendency for these cells to differentiate more towards effector CD8+ T cells, suggesting that TCF1 plays a pivotal role in the establishment of T cell exhaustion during chronic infection [56, 57]. PD-1 expression positively regulates TCF1 expression, indicating that PD-1 plays a crucial role in the functional regulation of exhausted CD8+T cells. Consequently, the expression levels of key transcription factors, such as T-bet, EOMES, and TCF1, in exhausted CD8+ T cells are closely linked to the expression of PD-1. This outcome suggests that PD-1 contributes to the molecular framework that governs the exhaustion process and the functional properties of CD8+ T cells during chronic infection [58]. The exact mechanisms by which these transcription factors influence CD8+ T cell function, as well as the interactions between PD-1 and transcription factors like T-bet, EOMES, and TCF1, remain unclear. Studies suggest that CTLA-4, secreted by T regulatory cells (Tregs), can disrupt the CD80/PD-L1 heterodimer on APCs, thereby increasing the availability of free PD-L1. This interaction may play a critical role in modulating immune responses and promoting immune dysfunction, particularly in chronic infections [44]. Therefore, elevated PD-L1 levels in OM may enhance PD-L1/PD-1 signaling, potentially regulating the expression of EOMES and contributing to the generation of exhausted CD8+ T cells. This signaling pathway may play a significant role in the immune dysfunction observed in chronic infections, such as OM [10].
Th17 and Treg cells, along with the Th17/Treg balance, are closely linked to the persistence of OM and bone destruction. Th17 cells, characterized by the surface marker RORγt, drive inflammation and suppress osteoclastogenesis. In contrast, Treg cells, marked by the transcription factor Foxp3, play a regulatory role by dampening inflammation—the deletion of Foxp3 results in a decrease in Treg cells. While Tregs primarily regulate the inflammatory response, they also promote osteoclastogenesis, contributing to the complex dynamics of bone remodeling in OM [59-61].
S. aureus can activate Treg cells, thereby suppressing the host immune response. Björkander et al. demonstrated that S. aureus stimulates T cells through superantigens, leading to a substantial secretion of cytokines. These cytokines, in turn, activate Treg cells, which contribute to the suppression of the immune response, facilitating the persistence of the infection [62].
In conclusion, various T cell subtypes have distinct roles in the progression of OM. CD4+ T cells, through the expression of CTLA-4, disrupt normal immune responses, highlighting the importance of investigating how S. aureus influences CTLA-4 expression and whether its virulence factors contribute to this modulation, which could reveal potential therapeutic targets. In OM, CD8+ T cells become exhausted, with key transcription factors such as T-bet, EOMES, and TCF1 playing crucial roles in this immune attenuation. Additionally, the opposing functions of Treg and Th17 cells warrant further exploration to understand better how to modulate their activity for therapeutic benefit in OM [10].
B cell
B lymphocytes are responsible for producing two distinct types of antibodies, ASN-1 and ASN-2, that play a critical role in neutralizing the virulence factors secreted by S. aureus. ASN-1 targets and neutralizes several key toxins, including alpha-hemolysin (Hla), Panton-Valentine leukocidin (PVL), leukocidin ED (LukED), and gamma-hemolysin. On the other hand, ASN-2 specifically neutralizes LukAB and LukGH, further protecting the host from the harmful effects of these toxins. These antibodies help reduce the damage caused by S. aureus, contributing to the body’s defense against the infection [63]. This expanded explanation clarifies the mechanism by which osteoclasts contribute to bone defects and highlights the role of macrophages in regulating osteoclast activity in inflammatory conditions, such as OM. It also highlights the significance of the RANKL/RANK pathway in maintaining bone homeostasis, particularly in disease states [64]. However, S. aureus employs immune evasion strategies to counteract humoral immunity. One such mechanism involves the virulence factor SpA, which disrupts antibody-mediated phagocytosis by binding to antibodies and simultaneously induces B cell apoptosis, thereby impairing the host’s immune defense [65-67]. The enzyme staphylokinase (Sak), produced by S. aureus, facilitates the cleavage and degradation of IgG antibodies, significantly impairing IgG-mediated phagocytosis by neutrophils and thereby enhancing the pathogen’s ability to evade the immune response [68]. Additionally, Pelzek et al. observed that while patients exhibited high levels of antibodies during the acute phase of S. aureus infection, this antibody response diminished by the 6-week follow-up visit. These findings suggest that S. aureus infections are insufficient to trigger robust secondary recall responses, failing to maintain elevated antibody levels over time. Based on this, although B cells can produce substantial antibodies against S. aureus virulence factors, the progression to chronic OM may be attributed to the inability of B cells to sustain antibody production at effective levels [69].
B cells are typically activated by lipopolysaccharides (LPS). However, lipoteichoic acid (LTA), a component of the cell membrane in gram-positive bacteria and structurally analogous to LPS, has been shown to inhibit B cell proliferation and interfere with LPS-induced B cell immune responses. While it remains challenging to establish whether LTA contributes directly to the immunosuppressive effects of S. aureus on B cells, its role offers a novel perspective for investigating the negative immunomodulatory influence of S. aureus in OM [70].
It has been demonstrated that in periodontitis, B cells contribute to inflammation and bone destruction by secreting pro-inflammatory cytokines, RANKL, MMPs, and autoantibodies (Abs). These mediators play a key role in intensifying the inflammatory response and facilitating tissue degradation [10, 71-73].
In conclusion, B cells, similar to other immune cells, are likely to play a significant role in the chronicity of OM and the associated bone destruction in S. aureus infections [10].
Chronic OM treatments and challenges
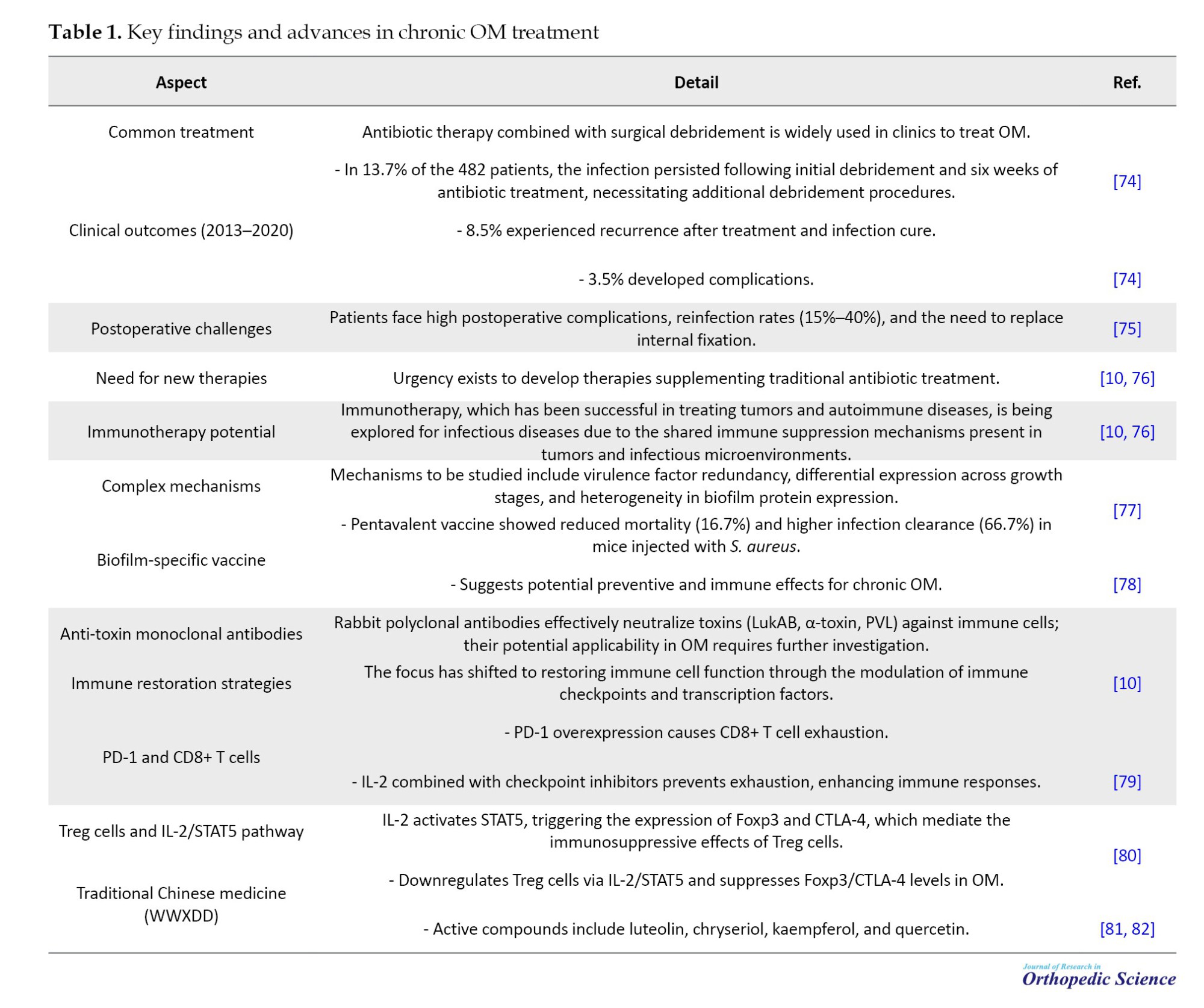
OM, a severe bone infection, presents significant clinical challenges despite advances in therapeutic strategies. Traditional approaches, such as antibiotic therapy combined with surgical debridement, often fall short due to high recurrence rates, postoperative complications, and reinfection. Emerging evidence highlights the potential of immunotherapy and alternative treatments, such as traditional Chinese medicine (TCM), to address these limitations. The following table summarizes key findings, clinical outcomes, and innovative therapeutic approaches for OM, drawing on recent studies and experimental data. Detailed information is presented in Table 1.
Bone resorption
Osteoclasts, originating from the monocyte/macrophage lineage, closely resemble macrophages within bone tissue [83]. Osteoclast precursor cells differentiate into mature osteoclasts, which serve as the primary mediators of bone resorption. This differentiation is largely regulated by the RANKL/RANK signaling pathway, particularly during infection. In post-traumatic OM, osteoclasts play a direct role in the development of bone defects. Additionally, macrophages contribute to osteoclast differentiation; notably, M1 macrophages secrete elevated levels of pro-inflammatory cytokines, such as TNF-α, IL-6, and IFN-γ, which promote osteoclastogenesis. This process stimulates the activation and function of osteoclasts, leading to increased bone resorption and the associated bone damage characteristic of OM [84, 85].
TNF-α is recognized as a key cytokine that regulates the RANKL/RANK signaling pathway. It not only modulates osteoblast function, thereby indirectly hindering bone formation, but also directly promotes osteoclast differentiation, thereby driving bone resorption [84, 86]. TRAF6 serves as a critical switch in the RANKL/RANK signaling pathway. TNF-α enhances the RANKL-TRAF6 signaling cascade by upregulating TRAF2, which in turn amplifies the activation of downstream signaling events. This interaction ultimately promotes osteoclastogenesis and bone resorption, contributing to bone degradation in inflammatory conditions such as OM [87]. The role of TNF-α in enhancing the RANKL/RANK signaling pathway remains a topic of debate. The majority of researchers argue that TNF-α acts synergistically with RANKL to enhance osteoclastogenesis, relying on RANKL to exert its effects. However, an opposing view suggests that TNF-α may independently influence this signaling pathway, potentially modulating osteoclast differentiation and function without the direct involvement of RANKL. Further studies are needed to clarify the precise interaction between TNF-α and RANKL in regulating osteoclastogenesis [88, 89].
Additionally, some studies have shown that TNF-α can regulate osteoclast growth independently of RANKL. This finding suggests that TNF-α may exert a direct effect on osteoclastogenesis through alternative signaling pathways or mechanisms that do not require the presence of RANKL. These findings highlight the complex and multifaceted role of TNF-α in bone metabolism, indicating that it may influence osteoclast differentiation and activity through both RANKL-dependent and RANKL-independent pathways. Further research is needed to understand better these interactions and their implications for bone diseases, such as OM [90]. Recent studies have revealed that TNF-α promotes the differentiation of osteoclast precursor cells through a mechanism involving sialylation. Sialic acid, a derivative of neuraminic acid, plays a critical role in osteoclastogenesis. TNF-α stimulates the differentiation of osteoclast precursors that are highly sialylated. However, this effect is significantly reduced when the sialylation process of precursor osteoclasts is disrupted. This result suggests that sialylation may be a key modulator in TNF-α-induced osteoclast differentiation, highlighting a potential therapeutic target for regulating osteoclastogenesis in conditions such as OM and other bone diseases [91].
Bone destruction
Th17 and Treg cells have opposing roles not only in regulating immune responses during inflammation but also in bone remodeling. Th17 cells primarily contribute to inflammation and bone degradation by producing pro-inflammatory cytokines, including IL-17, TNF-α, and IL-6, which promote osteoclastogenesis and enhance bone resorption. Conversely, Treg cells help control inflammation and protect against bone loss by releasing anti-inflammatory cytokines, including IL-10 and TGF-β. However, the balance between these two T cell subsets is crucial, as an imbalance can contribute to either excessive bone destruction or inadequate bone regeneration [10].
Treg cells inhibit osteoclast production and reduce bone destruction in OM, although the exact mechanisms behind this effect have remained controversial. Some studies suggest that Treg cells primarily exert their inhibitory effects on osteoclastogenesis by decreasing the secretion of pro-inflammatory cytokines, such as TGF-β, IL-4, and IL-10. These cytokines can modulate the activity of osteoclast precursor cells and disrupt the pro-osteoclastogenic signals, thereby limiting the differentiation and activity of osteoclasts. However, other research indicates that Treg cells may regulate osteoclastogenesis through different pathways, possibly involving direct cell-cell interactions or the modulation of other immune mediators [60]. The CTLA-4-mediated regulatory mechanism underscores the crucial role of Treg cells in maintaining immune homeostasis and controlling excessive bone destruction during chronic inflammation, such as OM [92]. However, some studies have demonstrated that Treg cells also promote osteoblastogenesis, thereby accelerating bone remodeling. Treg cells contribute to the regulation of osteoblast differentiation by secreting cytokines such as TGF-β and IL-10, which can stimulate the activity of osteoblasts.
Additionally, Treg cells may promote bone formation by enhancing the function of osteoblasts and supporting the deposition of new bone matrix. This dual role of Treg cells in both inhibiting osteoclastogenesis and promoting osteoblastogenesis underscores their complex involvement in bone remodeling, particularly in the context of OM, where the balance between bone resorption and formation needs to be tightly maintained [61]. The Wnt signaling pathway plays a critical role in regulating osteoblastogenesis by promoting the differentiation and proliferation of osteoblasts. Wnt10b, a ligand within this pathway, is a potent enhancer of osteoblast proliferation and bone formation. Under certain conditions, such as the presence of butyrate produced by Lactobacillus bacteria, Treg cells have been shown to regulate the activation of the Wnt10b promoter. This regulation ultimately leads to the increased expression of Wnt10b, which further enhances osteoblastogenesis. This finding suggests that Treg cells, through their ability to influence Wnt10b expression, contribute to bone remodeling by promoting osteoblast activity, which is essential for maintaining bone homeostasis in the context of inflammation and infection, such as OM [61].
IL-17 induces the secretion of pro-inflammatory cytokines, including IL-6, TNF-α, and IL-1, which further promote RANKL expression and activation in osteoblasts, thereby enhancing osteoclastogenesis. Additionally, Th17 cells themselves can secrete RANKL, contributing directly to osteoclast differentiation. This multifaceted role of Th17 cells in osteoclastogenesis links them to bone destruction, particularly in inflammatory conditions such as OM, where the balance between osteoblasts and osteoclasts is disrupted, leading to bone loss [93]. In conclusion, osteoblastogenesis is influenced by Th17 cells, which play a critical role in promoting bone destruction during inflammation. Through the secretion of IL-17 and other pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1, Th17 cells enhance RANKL expression in osteoblasts, which in turn stimulates osteoclastogenesis. Additionally, Th17 cells themselves can directly secrete RANKL, further contributing to bone resorption. The regulatory effect of Th17 cells on osteoblastogenesis highlights their role in bone destruction, particularly in inflammatory diseases such as OM, where the immune response leads to an imbalance between bone formation and resorption [10].
Th17 cells play a crucial role in promoting inflammation and bone destruction, while Treg cells have an opposing effect, suppressing inflammation and promoting bone formation. The balance between these two types of cells is tightly controlled under various physiological conditions. In states of intense inflammation, immune cells release elevated levels of cytokines such as IL-6, which drives the differentiation of Th17 cells through the activation of the STAT3 pathway. On the other hand, when IL-6 is absent, Th17 cell differentiation is inhibited. This shift in the balance between Th17 and Treg cells has a significant impact on both the inflammatory response and bone remodeling processes. Understanding this balance is essential for conditions like OM, where the inflammatory response not only affects immune activity but also disrupts bone homeostasis, leading to bone degradation [94-96].
Hypoxia-inducible factor α (HIF-1α) plays a crucial role in regulating the Th17/Treg ratio, particularly in hypoxic environments. Under low oxygen conditions, HIF1α is stabilized and activated, influencing the differentiation and function of various immune cells, including T cells. In hypoxia, HIF1α has been shown to promote the differentiation of Th17 cells while inhibiting the generation of Treg cells. This shift in the Th17/Treg balance can exacerbate inflammation and bone destruction, particularly in conditions like OM, where tissue hypoxia is often present. The modulation of HIF1α in these environments suggests its potential as a therapeutic target to manage inflammatory and immune responses in chronic infections [97]. HIF1α plays a critical role in regulating the Th17/Treg balance under hypoxic conditions. It enhances Th17 cell differentiation by upregulating the expression of RORγt, a key transcription factor driving Th17 differentiation. Simultaneously, HIF1α inhibits the expression of Foxp3, the master regulator of Treg cells, by promoting its degradation through the proteasomal pathway. This dual effect results in a relative increase in RORγt expression and a shift towards Th17 dominance. Such a shift in the Th17/Treg ratio under hypoxia contributes to increased inflammation and may exacerbate tissue damage in conditions like OM. Understanding this mechanism provides insight into potential therapeutic strategies for modulating immune responses in hypoxic environments [98].
OM and bone
OM is an invasive infectious condition that affects bone or bone marrow, leading to significant disruption of bone homeostasis and resulting in osteolysis. The classification of OM depends on the etiological agent (e.g. pyogenic, mycobacterial, or fungal infections) and the route of infection, which can be either hematogenous (via the bloodstream) or through direct extension from contiguous tissues. Additionally, OM can arise from infections in surrounding soft tissues that spread to the bone, direct inoculation during trauma or medical procedures, or specific anatomical regions such as the tibia or femur. The condition may also be categorized based on its duration, with acute and chronic forms of OM having distinct clinical features and treatment approaches [99].
Hematogenous OM is the most common form in children, accounting for a significant portion of cases. Approximately 60% of hematogenous OM cases are caused by S. aureus, a pathogen known for its virulence and ability to invade bone tissue. This form of OM typically results from the spread of bacteria through the bloodstream, which can reach bones, especially in growing children with highly vascularized bones. Early diagnosis and treatment are crucial for preventing complications such as bone deformities, chronic infections, and systemic spread [100]. OM has a higher incidence in males than females, particularly in children. This higher incidence in males may be attributed to factors such as hormonal differences, activity levels, and greater susceptibility to trauma. Despite advancements in treatment, approximately 30% of bone infections progress to chronic OM. Chronic OM can be challenging to treat and is associated with ongoing inflammation, bone destruction, and the potential for recurrent infections. Effective management requires timely intervention, appropriate antimicrobial therapy, and, in some cases, surgical debridement or stabilization to prevent further complications [101]. In adults, OM commonly affects individuals with underlying conditions such as diabetes, those who have experienced trauma, or those who have undergone orthopedic surgery. Diabetes increases the risk of OM due to impaired immune response, poor circulation, and the presence of neuropathy, which can lead to unnoticed injuries and infections. Trauma, such as fractures or open wounds, can directly introduce pathogens into the bone. At the same time, orthopedic surgeries, particularly joint replacements or internal fixation procedures, may provide a portal for bacteria to enter the bone, leading to infection. In these cases, timely diagnosis and targeted antimicrobial therapy are critical to prevent the progression to chronic OM, which can lead to significant morbidity [102]. Hip and knee replacement surgeries are among the most common causes of OM in adults. One of the major challenges in treating OM following these procedures is the formation of bacterial biofilms on the implanted foreign materials, such as prosthetic joints. Bacterial biofilms are clusters of bacteria encased in a protective matrix that adhere to surfaces, making them much more resistant to both the immune system and antibiotic treatments. This biofilm formation significantly complicates the treatment of infections, as the bacteria within the biofilm are less susceptible to standard antimicrobial therapies. As a result, surgical intervention, often including the removal of the infected implant, may be necessary in cases of chronic infection. Therefore, preventing biofilm formation and developing strategies to disrupt existing biofilms are key areas of focus in improving outcomes for patients with prosthetic-related OM [103].
Once the bone is infected, polymorphonuclear leukocytes (PMNs), also known as neutrophils, are recruited to the site of infection as part of the body’s innate immune response. These cells attempt to eliminate the infectious organisms by phagocytosing them. However, to effectively destroy the pathogens, activated PMNs release several highly reactive oxidants, including reactive oxygen species (ROS) and reactive nitrogen species (RNS), through processes such as the respiratory burst. While these oxidants play a critical role in pathogen destruction, they can also inadvertently cause damage to surrounding tissue. In the context of OM, the release of these reactive molecules can lead to the destruction of bone tissue, contributing to bone lysis and further exacerbating the infection. This immune-mediated damage to bone is a key factor in the progression of OM, as the bone’s integrity is compromised, and the healing process is impaired. Therefore, understanding and controlling the inflammatory response, including the activity of PMNs and the regulation of oxidative stress (OS), is essential in managing OM and preventing further tissue damage [104]. As the infection progresses, the pus produced by the pathogenic organisms spreads into the blood vessels within the bone, leading to significant alterations in blood flow. This disruption causes localized ischemia and the formation of devitalized, or necrotic, areas of bone. The lack of adequate blood supply to the affected bone tissue results in avascular necrosis, which is a hallmark of OM. In response to the infection and bone death, the body attempts to repair the damage by creating new bone around the necrotic area, a process known as involucrum formation. The involucrum is a layer of new bone that surrounds the dead bone, effectively encapsulating the sequestrum, or the dead bone, which becomes a reservoir for persistent infection. The presence of sequestrum contributes to the chronic nature of the infection, as it can serve as a nidus for ongoing bacterial activity, making it difficult for antibiotics and the immune system to eradicate the infection. This cycle of bone destruction, necrosis, and the body’s attempt at repair often leads to chronic OM, where infection and bone damage persist over time. Surgical removal of the sequestrum and proper antibiotic treatment are crucial for treating chronic OM and preventing further complications [104].
Chronic OM is often caused by persistent bacterial infection, with S. aureus being the most common pathogen responsible. This bacterium is particularly adept at evading the immune system and resisting treatment, which contributes to the persistence of the infection. The ability of S. aureus to form biofilms on bone and foreign materials (such as orthopedic implants) complicates treatment, as biofilms protect the bacteria from both the immune response and antibiotics. In chronic OM, S. aureus can establish a long-lasting infection by forming biofilms around areas of necrotic bone (sequestrum), making it difficult for antibiotics to penetrate and effectively eliminate the bacteria. The biofilm also creates a chronic inflammatory environment, where immune cells such as PMNs are activated but often unable to fully clear the infection. As a result, the infection persists, leading to ongoing bone destruction and the formation of new bone around the infected area (involucrum), which further complicates treatment and the healing process. Effective management of chronic OM requires a combination of surgical intervention to remove necrotic bone and infected tissue, along with prolonged antibiotic therapy that can penetrate biofilms and target the bacteria more effectively [100]. Although S. aureus has remained the most common cause of OM, other pathogens can also be involved, and the specific pathogens often vary with age, underlying conditions, and the route of infection. In children, Escherichia coli is a common causative organism of hematogenous OM, especially in cases associated with urinary tract infections or other systemic conditions. Other gram-negative bacteria, such as Klebsiella spp., Enterobacter spp., and Pseudomonas spp., are more frequently implicated in adults, particularly those with chronic medical conditions, such as diabetes, immunocompromised states, or post-surgical infections. Fungal, viral, and parasitic infections are far less common causes of OM. Fungal infections are typically seen in immunocompromised patients, while viral and parasitic infections causing OM are rare and generally secondary to other systemic infections. Anatomically, hematogenous OM tends to affect bones with ample blood supply, which can lead to more pronounced and rapid dissemination of pathogens. Commonly affected bones include the tibia, femur, humerus, vertebrae, and jaw. These bones are highly vascularized, making them more susceptible to bacterial seeding through the bloodstream during the acute phase of infection [3, 105].
S. aureus and osteoblasts
S. aureus can interact with osteoblasts not only on the cell surface but also intracellularly, due to its capability for internalization. This capability allows the bacteria to evade immune responses and persist within osteoblasts, potentially contributing to chronic infection and bone damage [106]. S. aureus contains various surface components, including cell wall peptidoglycans, lipoteichoic acid, and lipoproteins, which are collectively referred to as pathogen-associated molecular patterns (PAMPs). These PAMPs interact with osteoblasts, triggering the release of chemokines (such as CXCL2, CXCL8, CXCL10, CCL2, CCL3, and CCL5) and cytokines (including IL-1β, IL-18, and TNF-α). These molecules play a crucial role in recruiting and activating both innate immune cells (such as neutrophils and monocytes/macrophages) and adaptive immune cells (like lymphocytes), thereby enhancing the inflammatory response and promoting immune defense against the infection [107, 108]. S. aureus can interfere with bone formation by decreasing the expression of vital osteoblast markers necessary for their growth and proliferation. These markers include alkaline phosphatase, collagen type I, osteopontin, and osteocalcin, which are crucial for osteoblast function and bone matrix production. In addition, S. aureus stimulates osteoblasts to secrete soluble RANKL. This molecule plays a pivotal role in recruiting and activating osteoclasts, the cells responsible for bone resorption. As a result, the balance between bone formation and resorption is disrupted, leading to excessive bone loss and the promotion of inflammation-driven resorption, which contributes to skeletal damage. This mechanism highlights how S. aureus can not only impair osteoblast function but also drive osteoclast-mediated bone destruction, further complicating the inflammatory response in bone infections, such as OM [109, 110].
Ogawa was the first to present evidence showing that osteoblasts can internalize S. aureus [111]. The primary mechanism involves the interaction between fibronectin-binding proteins A and B (Fnbp A/B) on S. aureus and fibronectin, which acts as a mediator by connecting S. aureus to osteoblasts through the α5β1 integrin. This interaction facilitates the adhesion of the bacteria to osteoblasts, promoting infection and potentially influencing the immune response and bone remodeling processes [112].
By entering osteoblasts, S. aureus avoids detection by the immune system, promoting the persistence and spread of the infection. Inside the osteoblast, S. aureus resides within endolysosomal vesicles, where it can survive for prolonged periods [113-115].
S. aureus can persist for extended periods within osteoblasts, as evidenced by the presence of small colony variants. These variants exhibit an atypical, characteristic morphology that reduces their aggressiveness toward the host cell, thereby enhancing their long-term survival. However, despite these prolonged survival rates, the osteoblast’s safety is not guaranteed. Over time, the highly detrimental effects of PSMs on the cell membrane prove lethal, ultimately leading to the osteoblast’s death [115].
In response to S. aureus infection, osteoblasts do not remain inactive; rather, they actively release a variety of inflammatory mediators. These factors are part of the innate immune response and are crucial in initiating the body’s defense against the bacterial pathogen. Simultaneously, this response works in tandem with the adaptive immune system, particularly through the activation of Th1 lymphocytes, which further enhance the immune response and help coordinate the body’s efforts to combat the infection [116].
As previously mentioned, the conflict between S. aureus and osteoblasts is typically prolonged, with S. aureus emerging victorious, leading to subsequent bone loss. S. aureus is capable of inhibiting osteoblast activity and differentiation, while also preventing mineralization [117-120].
S. aureus can also induce osteoblast apoptosis through the activation of multiple pathways, all of which ultimately lead to the same outcome [121-123]. Undoubtedly, the death of osteoblasts is a hallmark event in OM caused by S. aureus, with dual consequences [124].
S. aureus and osteoclasts
Bone loss in OM occurs due to various factors and is not solely caused by the reduced function or death of osteoblasts. A key contributor to this process is the substantial increase in osteoclast differentiation and activity. Osteoblasts regulate osteoclastogenesis through the production of two important proteins: RANKL and OPG. RANKL, when bound to the RANK receptor on osteoclast precursors, triggers their differentiation into mature osteoclasts, which are responsible for bone resorption. On the other hand, OPG, which is produced by osteoblasts in a soluble form, acts as a decoy receptor. It binds to RANKL, preventing its interaction with RANK, and thus controls the differentiation and activation of osteoclasts. This balance between RANKL and OPG is crucial for bone homeostasis, and its disruption during OM leads to excessive osteoclast activity, contributing to bone destruction [125].
During S. aureus infection, osteoblasts elevate the production of RANKL while reducing the production of OPG, thereby enhancing osteoclastogenesis and accelerating bone resorption [110]. Research has also shown that osteoclasts infected with S. aureus produce substantial quantities of prostaglandin E2 (PGE2). This molecule binds to the EP4 receptor on osteoclasts, increasing RANKL production. The interaction between PGE2 and EP4 triggers a marked increase in RANKL levels, which in turn accelerates osteoclastogenesis, enhancing the differentiation and activation of osteoclasts. This mechanism highlights the role of PGE2 as a key mediator in the inflammatory process and bone resorption during S. aureus infection, further contributing to the pathological changes observed in OM [126, 127].
S. aureus (and or its components) can stimulate the release of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β. These cytokines play a key role in osteoclastogenesis by facilitating the differentiation and activation of osteoclasts from preosteoclasts [128, 129].
In animal models of induced OM, as well as in human bone samples and plasma from patients, these cytokines are found to be significantly elevated both in tissue and the circulatory system [130-132]. Osteoblasts secrete IL-6, as well as other mediators like CCL2 (monocyte chemotactic protein-1, MCP-1), CCL3 (macrophage inflammatory protein-1 alpha, MIP-1α), and CXCL-2 (MIP-2), in response to stimulation by S. aureus [133].
It is hypothesized that osteomacs, the macrophages present in bone, may act as additional contributors to the inflammation observed in OM [133].
A significant study demonstrated that the impact of S. aureus infection on osteoclast precursor cells derived from bone marrow is influenced by the stage of differentiation of these cells. When these precursor cells have already committed to the osteoclast lineage, the presence of RANKL promotes their differentiation into active osteoclasts, which are responsible for bone resorption. On the other hand, when the cells are still undifferentiated, RANKL cannot effectively trigger osteoclast differentiation. Instead, this scenario leads to the activation of M1 macrophages, which are known to contribute to the inflammatory process. This distinction highlights the complex relationship between infection, cell differentiation, and bone remodeling, suggesting that the stage of differentiation of osteoclast precursors plays a critical role in the inflammatory and bone-resorptive processes seen during S. aureus infection [9, 134].
S. aureus and osteocytes
Osteoblasts that lose their functional role become entrapped within the bone matrix they create and subsequently differentiate into osteocytes. These osteocytes, which have an irregular shape, extend their cytoplasmic processes into narrow channels known as canaliculi, forming a network that connects them with other osteocytes. This network facilitates the exchange of nutrients and waste products, which is crucial for maintaining the function and survival of osteocytes. Osteocytes play a crucial role in maintaining the integrity of the bone matrix, as they produce enzymes that help preserve the mineralized structure of the bone. Furthermore, osteocytes are capable of selectively removing minerals and restructuring the organic components of the bone, a process known as osteolysis, which plays a role in bone remodeling and maintaining bone health. This explanation highlights the dynamic roles of osteocytes in maintaining bone homeostasis and their active involvement in both bone maintenance and remodeling processes [3].
The involvement of bone remodeling in OM is still being studied. However, research has demonstrated an increase in metalloproteinase expression in osteocytes infected with S. aureus. This finding suggests that S. aureus infection may influence the process of osteolysis, potentially interfering with the overall process of bone remodeling. Osteolysis is crucial for maintaining the balance between bone formation and resorption, and its disruption in the context of OM may contribute to the structural and functional impairments observed in the bone tissue during infection [17].
Research has shown that human osteocyte-like cultures exposed to S. aureus exhibit a significant upregulation in the expression of several chemokines and cytokines, including CXCL9, CXCL10, and CXCL11. This observation suggests that osteocytes play an active role in attracting cytotoxic and or regulatory T-lymphocyte subsets to the sites of infection. By releasing these signaling molecules, osteocytes may contribute to the immune response and further influence the progression of the infection [17, 135].
Role of ROS in bone dynamics
ROS are molecules and free radicals produced as by-products of cellular processes, particularly oxidative phosphorylation in mitochondria. These species play crucial roles in regulating various physiological functions, including cell proliferation, metabolism, differentiation, and apoptosis. However, when their levels become dysregulated, often due to aging, inflammation, or age-related diseases like osteoarthritis, they contribute to OS and cellular damage. ROS also influences bone homeostasis, including the differentiation and activity of osteoclasts. The enzymes responsible for generating ROS, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, are pivotal in the regulation of bone resorption. Understanding the balance of ROS production and its impact on bone cells is crucial for developing therapeutic approaches to bone-related diseases. For a more detailed overview of ROS types and their roles in bone dynamics, refer to Table 2.

ROS signaling in bone remodeling
Bone homeostasis relies on a delicate balance between bone formation and resorption. With aging, this balance is often one of the first to be disrupted, as bone resorption tends to outpace bone formation [154]. ROS are key regulators of bone turnover, and extensive research has explored their role in influencing both bone formation and resorption [155]. Osteosclerosis can interfere with bone remodeling by creating an imbalance that promotes osteoclast activity, leading to the enhanced differentiation of pre-osteoclasts into mature osteoclasts. This disruption in the bone remodeling process may contribute to the development of metabolic bone diseases and skeletal disorders, including osteoporosis [156].
ROS can suppress or even completely halt the activity and differentiation of osteoblasts destined for apoptosis, while also affecting osteocytes. This disruption in osteoblast and osteocyte function further promotes osteoclastogenesis, leading to an imbalance in bone remodeling. ROS-induced alterations in osteoblast and osteocyte behavior can exacerbate the processes of bone resorption, contributing to conditions such as osteoporosis and other metabolic bone diseases [157-159].
Various factors, primarily secreted by osteoblasts and osteocytes, regulate the activity of osteoclasts and osteoblasts, thus influencing bone remodeling. Key factors are RANKL and OPG, as previously discussed. Their expression is closely influenced by an elevated oxidative state, resulting in the upregulation of RANKL and the downregulation of OPG. This process is mediated by the activation of protein kinases (such as ERK1/2, JNK) and/or other factors that modulate specific transcription factors [160-162].
OS inhibits osteoblast activation and reduces the production of OPG, thereby amplifying the effects of RANKL and stimulating osteoclast differentiation and activity. This disruption increases the RANKL/OPG ratio, a critical marker reflecting the degree of bone resorption [163]. An increase in the RANKL/OPG ratio indicates an imbalance that favors bone resorption over bone formation. This disruption is closely associated with the onset of various skeletal diseases, including different forms of osteoporosis and bone disorders secondary to inflammation, such as OM. Under normal conditions, the equilibrium between RANKL and OPG is tightly regulated to ensure a balanced process of bone formation and resorption. However, when this balance is disturbed and the RANKL/OPG ratio increases, bone resorption becomes dominant. This imbalance contributes to the degradation and thinning of bone tissue, which plays a central role in the development of diseases such as osteoporosis and OM. These conditions, often accompanied by inflammation, disrupt the physiological processes of bone remodeling, leading to weakened bones and an increased risk of fractures [3, 162, 164].
A self-perpetuating loop is created between OS and RANKL, where the excessive production of ROS inhibits osteoblast activation and OPG production. This condition increases RANKL, leading to enhanced osteoclastogenesis and subsequent bone resorption. RANKL, in turn, further promotes ROS production through the involvement of various intracellular signaling molecules such as TRAF6, Rac1, and NOX, amplifying the cycle and exacerbating bone degradation [165-167]. Furthermore, given the critical role of NOX in ROS production during osteoclast differentiation, the signaling cascade leading from RANK to ROS production can be outlined as follows: RANK activates TRAF6, which in turn activates Rac1. Rac1 then stimulates the NOX complex, resulting in the production of ROS. This pathway highlights the essential molecular interactions that contribute to ROS generation during osteoclastogenesis [168].
OS biomarkers in OM
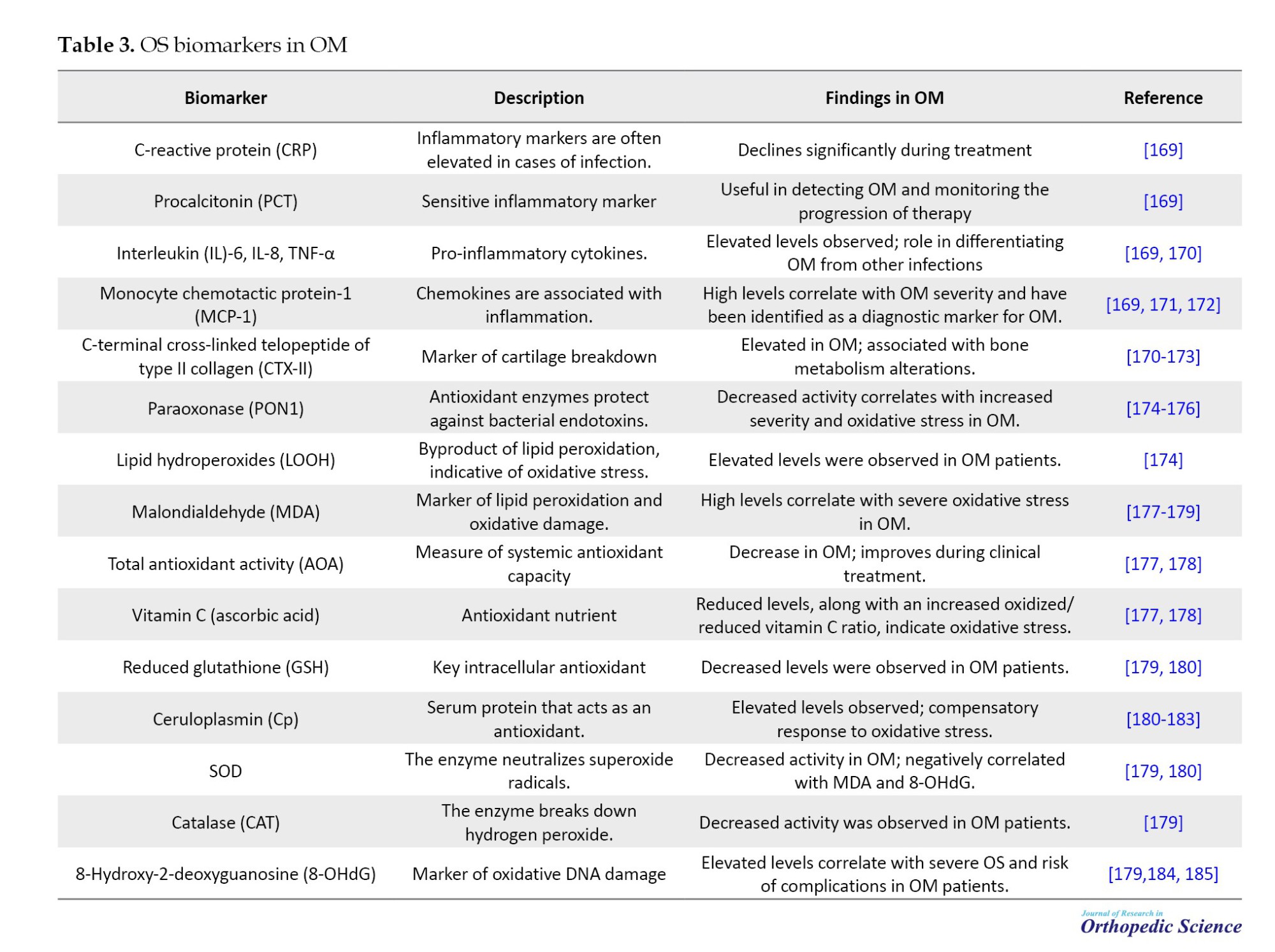
Oxidative stress plays a crucial role in the pathophysiology of OM, a severe musculoskeletal infection. OS arises from an imbalance between ROS and the body’s antioxidant defenses, contributing to tissue damage and the progression of disease. Multiple studies have highlighted the potential of OS biomarkers in diagnosing and monitoring OM. For example, paraoxonase-1 activity, serum lipid hydroperoxides, and 8-hydroxy-2-deoxyguanosine (8-OHdG) levels have been shown to correlate with the severity of oxidative damage in OM patients. These markers offer insight into disease status and may inform therapeutic strategies aimed at mitigating oxidative damage and enhancing patient outcomes. For detailed findings on oxidative stress biomarkers in OM, refer to Table 3.
Conclusion
OM represents a significant challenge in both clinical and molecular research due to its complex pathophysiology and impact on bone health. The molecular mechanisms underlying the infection involve a coordinated response between the immune system and bone cells, resulting in inflammation, bone resorption, and impaired bone healing. Understanding these molecular pathways is essential for identifying novel therapeutic targets that could enhance the treatment of OM. Further research is needed to explore the cellular interactions involved and to develop more effective strategies for preventing bone destruction and promoting bone regeneration in affected individuals.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization and supervision: Abolfazl Bagherifard and Hooman Yahyazedeh; Validation : Hooman Yahyazedeh and Khatere Mokhtari; Writing the original draft: Khatere Mokhtari and Abolfazl Bagherifard; Investigation, review and editing: All authors.
Conflict of interest
The authors declared no conflict of interest.
References
Bone
Bone is a highly dynamic tissue that continuously remodels itself to preserve homeostasis. This process is regulated by two distinct types of cells: Osteoblasts, which arise from mesenchymal stem cells and are responsible for forming new bone, and osteoclasts, which originate from hematopoietic precursors and mediate bone resorption. The differentiation of osteoclasts from monocytes is regulated by two cytokines: M-CSF and receptor activator of NF-κB ligand (RANKL). When these cytokines bind to their respective receptors on monocytes, they initiate the differentiation process into osteoclasts [1-3]. In the body, ongoing bone remodeling helps repair microfractures and compensates for wear caused by biomechanical stress, typically resulting in complete renewal within 7 to 10 months [4-6]. The skeletal system is highly sensitive to both local and systemic signals. Several diseases are characterized by heightened bone resorption, which may be associated with hormonal imbalances and aging in certain cases [7, 8].
Staphylococcus aureus
S. aureus is the leading pathogen responsible for osteomyelitis (OM) and is one of the most common microorganisms implicated in persistent infections [9, 10]. S. aureus interferes with immune cell functions and disrupts the equilibrium between osteoclasts and osteoblasts, leading to sustained infection and continuous bone degradation, which are critical factors in the progression of chronic OM [9]. S. aureus promotes chronic infection and bone destruction through several key mechanisms. It secretes virulence factors that enhance bacterial colonization, induce the death of immune cells, and alter the immune environment, allowing the bacteria to evade the immune system. These virulence factors are also associated with the formation of abscesses in OM. Additionally, S. aureus forms biofilms in response to vascular damage and reduced oxygen levels, creating a protective barrier that prevents immune cells from contacting the pathogen, impairs neutrophil activation, and encourages macrophage differentiation. The pathogen also adjusts its metabolism during infection, enabling it to compete with immune cells for energy and survive. Finally, S. aureus can survive within immune cells by escaping the phagosome and continuing to replicate within the cell [10-16].
Molecular insight into S. aureus
S. aureus infiltrates the cortical bone, infecting osteocytes. In response, these infected osteocytes release elevated levels of chemokines, including CCL5, CXCL1, CXCL8, CXCL9, and CXCL11. Neutrophils and T lymphocytes are activated by these chemokines, which in turn drive inflammation and contribute to the destruction of bone tissue. This immune response, while aimed at controlling the infection, also exacerbates bone damage, leading to the progression of OM [17]. S. aureus can also survive within osteoblasts and cause morphological changes or even cell death. Infected osteoblasts release higher levels of inflammatory factors, which promote the migration of immune cells and stimulate osteoclastogenesis [9, 10, 18].
Neutrophils
At the early stage of infection, neutrophils are the first immune cells to become activated and carry out their defensive functions at the site of infection [19]. However, during S. aureus-induced OM, key neutrophil functions—including chemotactic migration, activation, and phagocytic killing—are disrupted. This condition hinders the formation of neutrophil extracellular traps (NETs) and diminishes their ability to eliminate bacteria. Moreover, neutrophils undergo modifications during the infection that contribute to tissue damage and facilitate bacterial survival and spread at the infection site. Initially, host cells release higher levels of chemokines to attract neutrophils to the infection site, where they perform bactericidal functions. However, S. aureus secretes virulence factors such as chronic inflammatory proteins (CHIPs) and the FPRL1 inhibitory protein (FLIPr), which interfere with neutrophil chemotaxis in OM. CHIPs reduce the production of chemokines and impair neutrophil migration toward the site of infection, thereby hindering the host’s ability to combat bacterial invasion effectively [20, 21].
Neutrophil activation normally depends on the recognition of pathogen-specific receptors. However, Ssl3—a virulence factor secreted by S. aureus and part of the Ssl protein family—interferes with this process by inhibiting pathogen recognition. SSL3 inhibits neutrophil activation by competitively binding to TLR2, thus impairing an effective immune response. Additionally, the virulence factor CHIPs competes with complement C5a for binding, preventing C5a from activating neutrophils via its receptor [11, 22, 23]. In response to immune challenges, neutrophils release inhibitory proteins, such as neutrophil elastase, proteinase-3, and cathepsin G, to degrade CHIPs. However, S. aureus counteracts this defense mechanism by producing extracellular adherence protein (Eap) and its homologs, EapH1 and EapH2. These proteins inhibit the activity of neutrophil elastases, effectively protecting the bacteria from immune clearance and allowing them to persist in the infection site [24]. Moreover, in chronic infections, some neutrophils express MHC class II molecules, allowing them to present antigens to T cells. Research has shown that under conditions of persistent infection, these neutrophils can participate in antigen presentation and activate T cells, thus playing a role in the adaptive immune response [25].
A hallmark characteristic of S. aureus in OM is its ability to form biofilms on implant surfaces. When biofilms are present, the bacteria are protected from immune surveillance, unlike planktonic bacteria, which are rapidly detected and cleared by immune cells in the absence of biofilm formation. Biofilm formation enables bacteria to evade immune responses and persist at the site of infection. However, when planktonic bacteria adhere to implant surfaces, such as internal fixation devices (e.g. plates, screws), biofilms are formed, creating a protective niche that shields the bacteria from immune detection and promotes their persistence [13]. Immature biofilms do not provide a strong barrier and can still be penetrated by fully activated neutrophils, enabling them to perform their immune functions. However, as the biofilm matures, its ability to act as a physical shield against immune cells increases significantly. Extracellular polymeric substances (EPS), a vital component of biofilms, play a pivotal role in immunosuppression. EPS hinders neutrophil recognition of bacteria by redirecting their attention from the pathogen to the biofilm itself, thereby aiding the pathogen in evading the host’s immune system and promoting its persistence at the infection site [26, 27].
The ability of S. aureus to persistently survive within immune cells is a critical factor that impedes neutrophils from effectively performing phagocytosis and eliminating pathogens. S. aureus disrupts the function of neutrophil phagosomes, facilitating its survival within these immune cells [28-30]. In OM, S. aureus likely survives within the phagosome by disrupting the expression of LC3-modified proteins. Understanding the mechanisms by which S. aureus regulates LC3 expression in this context is essential. Targeting and inhibiting these pathways could offer a novel approach to restore phagosome function and enhance the ability of immune cells to eliminate pathogens efficiently [31].
The primary function of NETs is to capture and eliminate S. aureus at the site of infection. However, this process may result in damage to endothelial cells and other adjacent tissues, potentially contributing to additional tissue injury and complications [32]. Research has demonstrated that histone proteins mediate platelet activation through TLR4 and TLR2, thereby promoting blood coagulation and thrombin formation, a process linked to platelet-rich microthrombosis in sepsis models. Thus, histones found in NETs may play a pivotal role in the development of microthrombosis in chronic OM. This microthrombosis could impede tissue repair and create an environment conducive to the growth of S. aureus. Consequently, NETs not only serve to capture pathogens but also substantially contribute to tissue damage in chronic OM [33].
Macrophages
During the initial phase of infection, circulating monocytes differentiate into macrophages in response to the influence of inflammatory cytokines. M1 macrophages play a key role in pathogen elimination by releasing lysosomal enzymes and enhancing inflammation through the secretion of pro-inflammatory cytokines. In contrast, M2 macrophages secrete anti-inflammatory cytokines to modulate the inflammatory response and produce growth factors, such as platelet-derived growth factor and fibroblast growth factor, which contribute to tissue repair and healing [34]. In chronic OM, there is an imbalance and dysregulation in the interconversion and proportion of M1 and M2 macrophages. The prolonged presence of M1 macrophages contributes to an exaggerated inflammatory response, which can potentially cause significant tissue damage. On the other hand, an overproduction of M2 macrophages can impair effective phagocytosis and cytotoxic activity, thereby favoring biofilm formation and enhancing bacterial resistance, which hinders the resolution of infection [35].
These findings suggest that the STAT3/STAT6 pathway may impede the pathogen-clearing function of macrophages, thereby facilitating the persistence of OM. Additionally, IL-10 has been shown to activate the STAT3 pathway, which further suppresses the macrophage immune response. As a result, targeting this signaling pathway could offer a potential immunoregulatory strategy for treating chronic OM [36-38]. The biofilm formed by S. aureus also plays a crucial role in modulating macrophage polarization [39, 40].
In chronic OM, the migration of macrophages is regulated by the NF-κB/TWIST1 signaling pathway. This pathway plays a crucial role in modulating the movement and activation of macrophages, which are essential for the inflammatory response and tissue remodeling during the progression of the infection [35]. Additionally, TWIST1 enhances the expression of matrix metalloproteinases (MMP9 and MMP3), which facilitate macrophage migration by breaking down extracellular matrix components at the infection site. This degradation allows macrophages to move more effectively towards areas of infection, thereby contributing to the inflammatory response and tissue remodeling seen in chronic OM [41].
The PI3K/Akt-Beclin signaling pathway plays a crucial role in regulating macrophage autophagy, pathogen phagocytosis, and the NF-κB-mediated inflammatory response in S. aureus-induced OM. Recent studies have shown that inhibiting PI3K impairs macrophage autophagy, thereby reducing their ability to phagocytose S. aureus [42] effectively.
T cells
Upon encountering an antigen, T lymphocytes differentiate into two main subsets: Helper T cells (CD4+ T cells) and cytotoxic T cells (CD8+ T cells). CD4+ T cells bolster immune responses by releasing a variety of inflammatory mediators that activate other immune cells and regulate inflammation by controlling the proliferation of certain immune cell types. In contrast, CD8+ T cells target and eliminate pathogens directly by secreting cytotoxic proteins and cytokines, such as perforin and granzyme [26]. Dendritic cells play a crucial role in activating T cells through the presentation of antigens. However, leukocidin AB (LukAB), a virulence factor secreted by S. aureus, disrupts this process by inducing the death of dendritic cells, which are crucial for effective antigen presentation and the subsequent initiation of immune responses. This interference by LukAB undermines the immune system’s ability to recognize and respond to the pathogen [43] properly. CTLA-4, an inhibitory molecule predominantly expressed on T cells, functions by binding to CD80/CD86 molecules present on antigen-presenting cells (APCs). This binding prevents the interaction between CD28 on T cells and these costimulatory molecules, thereby inhibiting the activation of T cells. As a result, the immune response is compromised, impeding the clearance of pathogens and allowing the infection to persist [26, 44]. As OM transitions from the acute to the chronic phase, the functionality of CD8+ T cells is significantly impaired, causing them to contribute more to the pathogenesis of chronic OM rather than assisting in the elimination of pathogens. This dysfunction in CD8+ T cell activity enables the infection to persist and worsen over time [10].
Studies have demonstrated that in chronic OM, the cytotoxic function of CD8+ T cells is notably compromised due to prolonged exposure to elevated antigen levels and inflammatory mediators. These T cells, which exhibit immunosuppressive characteristics, are classified as exhausted CD8+ T cells [44]. Exhausted CD8+ T cells show a significant decline in the production of crucial cytokines, such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-12, IL-18, and IL-10. Additionally, the synthesis of cytotoxic proteins, including granzyme and perforin, is impaired, thereby compromising their ability to target and eliminate pathogens efficiently [45]. Importantly, exhausted CD8+ T cells express various inhibitory receptors, including PD-1, LAG-3, 2B4, CD160, TIM-3, and TIGHT, which contribute to their functional impairment and inability to combat pathogens [45-47] effectively. During chronic infections, inhibitory receptors continue to exert immunosuppressive effects. Although the impact of each receptor individually may be modest, exhausted T cells typically co-express several receptors, such as PD-1 and LAG-3. This co-expression leads to a synergistic effect, resulting in a more substantial immunosuppressive response than any single receptor could induce on its own [48]. Moreover, the metabolic profile of exhausted CD8+ T cells changes to adapt to the environment of persistent infection. These alterations enable the cells to survive and function under chronic stress conditions, though they may impair their full effector functions and contribute to immune dysfunction [49-53]. Recent studies have highlighted a notable distinction in the expression of transcription factors between normal effector CD8+ T cells and their exhausted counterparts. Transcription factors such as T-bet, EOMES, and TCF1 have gained recognition for their pivotal role in driving the functional shift of CD8+ T cells toward exhaustion. These factors are integral to regulating CD8+T cell functionality and facilitate their transition into an exhausted state, particularly in the context of chronic infections [10].
T-bet and EOMES are essential transcription factors that regulate the differentiation of naive CD8+ T cells. When T-bet expression surpasses that of EOMES, CD8+ T cells are directed towards differentiating into terminal effector cells, which are critical for executing immune responses. In contrast, when EOMES expression surpasses that of T-bet, naive CD8+ T cells are more likely to differentiate into memory T cells. However, sustained high expression of EOMES is associated with the progressive exhaustion of CD8+ T cells, leading to diminished immune function in chronic infections [54]. Some researchers propose that the sustained high expression of EOMES may be linked to elevated levels of PD-1, a marker known to be upregulated during chronic OM. This outcome suggests that the overexpression of EOMES may contribute to the exhaustion of CD8+ T cells by promoting PD-1 expression, which in turn impairs T cell function and supports the persistence of the infection [48, 55].
TCF1, a transcription factor exclusive to T cells, plays a pivotal role in the differentiation and sustained function of exhausted CD8+ T cells. During the initial stages of chronic infection, TCF1 is co-expressed with CXCL5 in CD8+ T cells. However, as the infection progresses, these cells progressively adopt exhausted characteristics, leading to a decline in their effector functions and contributing to the persistence of the infection. When TCF1 is knocked down in CD8+ T cells, there is a tendency for these cells to differentiate more towards effector CD8+ T cells, suggesting that TCF1 plays a pivotal role in the establishment of T cell exhaustion during chronic infection [56, 57]. PD-1 expression positively regulates TCF1 expression, indicating that PD-1 plays a crucial role in the functional regulation of exhausted CD8+T cells. Consequently, the expression levels of key transcription factors, such as T-bet, EOMES, and TCF1, in exhausted CD8+ T cells are closely linked to the expression of PD-1. This outcome suggests that PD-1 contributes to the molecular framework that governs the exhaustion process and the functional properties of CD8+ T cells during chronic infection [58]. The exact mechanisms by which these transcription factors influence CD8+ T cell function, as well as the interactions between PD-1 and transcription factors like T-bet, EOMES, and TCF1, remain unclear. Studies suggest that CTLA-4, secreted by T regulatory cells (Tregs), can disrupt the CD80/PD-L1 heterodimer on APCs, thereby increasing the availability of free PD-L1. This interaction may play a critical role in modulating immune responses and promoting immune dysfunction, particularly in chronic infections [44]. Therefore, elevated PD-L1 levels in OM may enhance PD-L1/PD-1 signaling, potentially regulating the expression of EOMES and contributing to the generation of exhausted CD8+ T cells. This signaling pathway may play a significant role in the immune dysfunction observed in chronic infections, such as OM [10].
Th17 and Treg cells, along with the Th17/Treg balance, are closely linked to the persistence of OM and bone destruction. Th17 cells, characterized by the surface marker RORγt, drive inflammation and suppress osteoclastogenesis. In contrast, Treg cells, marked by the transcription factor Foxp3, play a regulatory role by dampening inflammation—the deletion of Foxp3 results in a decrease in Treg cells. While Tregs primarily regulate the inflammatory response, they also promote osteoclastogenesis, contributing to the complex dynamics of bone remodeling in OM [59-61].
S. aureus can activate Treg cells, thereby suppressing the host immune response. Björkander et al. demonstrated that S. aureus stimulates T cells through superantigens, leading to a substantial secretion of cytokines. These cytokines, in turn, activate Treg cells, which contribute to the suppression of the immune response, facilitating the persistence of the infection [62].
In conclusion, various T cell subtypes have distinct roles in the progression of OM. CD4+ T cells, through the expression of CTLA-4, disrupt normal immune responses, highlighting the importance of investigating how S. aureus influences CTLA-4 expression and whether its virulence factors contribute to this modulation, which could reveal potential therapeutic targets. In OM, CD8+ T cells become exhausted, with key transcription factors such as T-bet, EOMES, and TCF1 playing crucial roles in this immune attenuation. Additionally, the opposing functions of Treg and Th17 cells warrant further exploration to understand better how to modulate their activity for therapeutic benefit in OM [10].
B cell
B lymphocytes are responsible for producing two distinct types of antibodies, ASN-1 and ASN-2, that play a critical role in neutralizing the virulence factors secreted by S. aureus. ASN-1 targets and neutralizes several key toxins, including alpha-hemolysin (Hla), Panton-Valentine leukocidin (PVL), leukocidin ED (LukED), and gamma-hemolysin. On the other hand, ASN-2 specifically neutralizes LukAB and LukGH, further protecting the host from the harmful effects of these toxins. These antibodies help reduce the damage caused by S. aureus, contributing to the body’s defense against the infection [63]. This expanded explanation clarifies the mechanism by which osteoclasts contribute to bone defects and highlights the role of macrophages in regulating osteoclast activity in inflammatory conditions, such as OM. It also highlights the significance of the RANKL/RANK pathway in maintaining bone homeostasis, particularly in disease states [64]. However, S. aureus employs immune evasion strategies to counteract humoral immunity. One such mechanism involves the virulence factor SpA, which disrupts antibody-mediated phagocytosis by binding to antibodies and simultaneously induces B cell apoptosis, thereby impairing the host’s immune defense [65-67]. The enzyme staphylokinase (Sak), produced by S. aureus, facilitates the cleavage and degradation of IgG antibodies, significantly impairing IgG-mediated phagocytosis by neutrophils and thereby enhancing the pathogen’s ability to evade the immune response [68]. Additionally, Pelzek et al. observed that while patients exhibited high levels of antibodies during the acute phase of S. aureus infection, this antibody response diminished by the 6-week follow-up visit. These findings suggest that S. aureus infections are insufficient to trigger robust secondary recall responses, failing to maintain elevated antibody levels over time. Based on this, although B cells can produce substantial antibodies against S. aureus virulence factors, the progression to chronic OM may be attributed to the inability of B cells to sustain antibody production at effective levels [69].
B cells are typically activated by lipopolysaccharides (LPS). However, lipoteichoic acid (LTA), a component of the cell membrane in gram-positive bacteria and structurally analogous to LPS, has been shown to inhibit B cell proliferation and interfere with LPS-induced B cell immune responses. While it remains challenging to establish whether LTA contributes directly to the immunosuppressive effects of S. aureus on B cells, its role offers a novel perspective for investigating the negative immunomodulatory influence of S. aureus in OM [70].
It has been demonstrated that in periodontitis, B cells contribute to inflammation and bone destruction by secreting pro-inflammatory cytokines, RANKL, MMPs, and autoantibodies (Abs). These mediators play a key role in intensifying the inflammatory response and facilitating tissue degradation [10, 71-73].
In conclusion, B cells, similar to other immune cells, are likely to play a significant role in the chronicity of OM and the associated bone destruction in S. aureus infections [10].
Chronic OM treatments and challenges
OM, a severe bone infection, presents significant clinical challenges despite advances in therapeutic strategies. Traditional approaches, such as antibiotic therapy combined with surgical debridement, often fall short due to high recurrence rates, postoperative complications, and reinfection. Emerging evidence highlights the potential of immunotherapy and alternative treatments, such as traditional Chinese medicine (TCM), to address these limitations. The following table summarizes key findings, clinical outcomes, and innovative therapeutic approaches for OM, drawing on recent studies and experimental data. Detailed information is presented in Table 1.
Bone resorption
Osteoclasts, originating from the monocyte/macrophage lineage, closely resemble macrophages within bone tissue [83]. Osteoclast precursor cells differentiate into mature osteoclasts, which serve as the primary mediators of bone resorption. This differentiation is largely regulated by the RANKL/RANK signaling pathway, particularly during infection. In post-traumatic OM, osteoclasts play a direct role in the development of bone defects. Additionally, macrophages contribute to osteoclast differentiation; notably, M1 macrophages secrete elevated levels of pro-inflammatory cytokines, such as TNF-α, IL-6, and IFN-γ, which promote osteoclastogenesis. This process stimulates the activation and function of osteoclasts, leading to increased bone resorption and the associated bone damage characteristic of OM [84, 85].
TNF-α is recognized as a key cytokine that regulates the RANKL/RANK signaling pathway. It not only modulates osteoblast function, thereby indirectly hindering bone formation, but also directly promotes osteoclast differentiation, thereby driving bone resorption [84, 86]. TRAF6 serves as a critical switch in the RANKL/RANK signaling pathway. TNF-α enhances the RANKL-TRAF6 signaling cascade by upregulating TRAF2, which in turn amplifies the activation of downstream signaling events. This interaction ultimately promotes osteoclastogenesis and bone resorption, contributing to bone degradation in inflammatory conditions such as OM [87]. The role of TNF-α in enhancing the RANKL/RANK signaling pathway remains a topic of debate. The majority of researchers argue that TNF-α acts synergistically with RANKL to enhance osteoclastogenesis, relying on RANKL to exert its effects. However, an opposing view suggests that TNF-α may independently influence this signaling pathway, potentially modulating osteoclast differentiation and function without the direct involvement of RANKL. Further studies are needed to clarify the precise interaction between TNF-α and RANKL in regulating osteoclastogenesis [88, 89].
Additionally, some studies have shown that TNF-α can regulate osteoclast growth independently of RANKL. This finding suggests that TNF-α may exert a direct effect on osteoclastogenesis through alternative signaling pathways or mechanisms that do not require the presence of RANKL. These findings highlight the complex and multifaceted role of TNF-α in bone metabolism, indicating that it may influence osteoclast differentiation and activity through both RANKL-dependent and RANKL-independent pathways. Further research is needed to understand better these interactions and their implications for bone diseases, such as OM [90]. Recent studies have revealed that TNF-α promotes the differentiation of osteoclast precursor cells through a mechanism involving sialylation. Sialic acid, a derivative of neuraminic acid, plays a critical role in osteoclastogenesis. TNF-α stimulates the differentiation of osteoclast precursors that are highly sialylated. However, this effect is significantly reduced when the sialylation process of precursor osteoclasts is disrupted. This result suggests that sialylation may be a key modulator in TNF-α-induced osteoclast differentiation, highlighting a potential therapeutic target for regulating osteoclastogenesis in conditions such as OM and other bone diseases [91].
Bone destruction
Th17 and Treg cells have opposing roles not only in regulating immune responses during inflammation but also in bone remodeling. Th17 cells primarily contribute to inflammation and bone degradation by producing pro-inflammatory cytokines, including IL-17, TNF-α, and IL-6, which promote osteoclastogenesis and enhance bone resorption. Conversely, Treg cells help control inflammation and protect against bone loss by releasing anti-inflammatory cytokines, including IL-10 and TGF-β. However, the balance between these two T cell subsets is crucial, as an imbalance can contribute to either excessive bone destruction or inadequate bone regeneration [10].
Treg cells inhibit osteoclast production and reduce bone destruction in OM, although the exact mechanisms behind this effect have remained controversial. Some studies suggest that Treg cells primarily exert their inhibitory effects on osteoclastogenesis by decreasing the secretion of pro-inflammatory cytokines, such as TGF-β, IL-4, and IL-10. These cytokines can modulate the activity of osteoclast precursor cells and disrupt the pro-osteoclastogenic signals, thereby limiting the differentiation and activity of osteoclasts. However, other research indicates that Treg cells may regulate osteoclastogenesis through different pathways, possibly involving direct cell-cell interactions or the modulation of other immune mediators [60]. The CTLA-4-mediated regulatory mechanism underscores the crucial role of Treg cells in maintaining immune homeostasis and controlling excessive bone destruction during chronic inflammation, such as OM [92]. However, some studies have demonstrated that Treg cells also promote osteoblastogenesis, thereby accelerating bone remodeling. Treg cells contribute to the regulation of osteoblast differentiation by secreting cytokines such as TGF-β and IL-10, which can stimulate the activity of osteoblasts.
Additionally, Treg cells may promote bone formation by enhancing the function of osteoblasts and supporting the deposition of new bone matrix. This dual role of Treg cells in both inhibiting osteoclastogenesis and promoting osteoblastogenesis underscores their complex involvement in bone remodeling, particularly in the context of OM, where the balance between bone resorption and formation needs to be tightly maintained [61]. The Wnt signaling pathway plays a critical role in regulating osteoblastogenesis by promoting the differentiation and proliferation of osteoblasts. Wnt10b, a ligand within this pathway, is a potent enhancer of osteoblast proliferation and bone formation. Under certain conditions, such as the presence of butyrate produced by Lactobacillus bacteria, Treg cells have been shown to regulate the activation of the Wnt10b promoter. This regulation ultimately leads to the increased expression of Wnt10b, which further enhances osteoblastogenesis. This finding suggests that Treg cells, through their ability to influence Wnt10b expression, contribute to bone remodeling by promoting osteoblast activity, which is essential for maintaining bone homeostasis in the context of inflammation and infection, such as OM [61].
IL-17 induces the secretion of pro-inflammatory cytokines, including IL-6, TNF-α, and IL-1, which further promote RANKL expression and activation in osteoblasts, thereby enhancing osteoclastogenesis. Additionally, Th17 cells themselves can secrete RANKL, contributing directly to osteoclast differentiation. This multifaceted role of Th17 cells in osteoclastogenesis links them to bone destruction, particularly in inflammatory conditions such as OM, where the balance between osteoblasts and osteoclasts is disrupted, leading to bone loss [93]. In conclusion, osteoblastogenesis is influenced by Th17 cells, which play a critical role in promoting bone destruction during inflammation. Through the secretion of IL-17 and other pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1, Th17 cells enhance RANKL expression in osteoblasts, which in turn stimulates osteoclastogenesis. Additionally, Th17 cells themselves can directly secrete RANKL, further contributing to bone resorption. The regulatory effect of Th17 cells on osteoblastogenesis highlights their role in bone destruction, particularly in inflammatory diseases such as OM, where the immune response leads to an imbalance between bone formation and resorption [10].
Th17 cells play a crucial role in promoting inflammation and bone destruction, while Treg cells have an opposing effect, suppressing inflammation and promoting bone formation. The balance between these two types of cells is tightly controlled under various physiological conditions. In states of intense inflammation, immune cells release elevated levels of cytokines such as IL-6, which drives the differentiation of Th17 cells through the activation of the STAT3 pathway. On the other hand, when IL-6 is absent, Th17 cell differentiation is inhibited. This shift in the balance between Th17 and Treg cells has a significant impact on both the inflammatory response and bone remodeling processes. Understanding this balance is essential for conditions like OM, where the inflammatory response not only affects immune activity but also disrupts bone homeostasis, leading to bone degradation [94-96].
Hypoxia-inducible factor α (HIF-1α) plays a crucial role in regulating the Th17/Treg ratio, particularly in hypoxic environments. Under low oxygen conditions, HIF1α is stabilized and activated, influencing the differentiation and function of various immune cells, including T cells. In hypoxia, HIF1α has been shown to promote the differentiation of Th17 cells while inhibiting the generation of Treg cells. This shift in the Th17/Treg balance can exacerbate inflammation and bone destruction, particularly in conditions like OM, where tissue hypoxia is often present. The modulation of HIF1α in these environments suggests its potential as a therapeutic target to manage inflammatory and immune responses in chronic infections [97]. HIF1α plays a critical role in regulating the Th17/Treg balance under hypoxic conditions. It enhances Th17 cell differentiation by upregulating the expression of RORγt, a key transcription factor driving Th17 differentiation. Simultaneously, HIF1α inhibits the expression of Foxp3, the master regulator of Treg cells, by promoting its degradation through the proteasomal pathway. This dual effect results in a relative increase in RORγt expression and a shift towards Th17 dominance. Such a shift in the Th17/Treg ratio under hypoxia contributes to increased inflammation and may exacerbate tissue damage in conditions like OM. Understanding this mechanism provides insight into potential therapeutic strategies for modulating immune responses in hypoxic environments [98].
OM and bone
OM is an invasive infectious condition that affects bone or bone marrow, leading to significant disruption of bone homeostasis and resulting in osteolysis. The classification of OM depends on the etiological agent (e.g. pyogenic, mycobacterial, or fungal infections) and the route of infection, which can be either hematogenous (via the bloodstream) or through direct extension from contiguous tissues. Additionally, OM can arise from infections in surrounding soft tissues that spread to the bone, direct inoculation during trauma or medical procedures, or specific anatomical regions such as the tibia or femur. The condition may also be categorized based on its duration, with acute and chronic forms of OM having distinct clinical features and treatment approaches [99].
Hematogenous OM is the most common form in children, accounting for a significant portion of cases. Approximately 60% of hematogenous OM cases are caused by S. aureus, a pathogen known for its virulence and ability to invade bone tissue. This form of OM typically results from the spread of bacteria through the bloodstream, which can reach bones, especially in growing children with highly vascularized bones. Early diagnosis and treatment are crucial for preventing complications such as bone deformities, chronic infections, and systemic spread [100]. OM has a higher incidence in males than females, particularly in children. This higher incidence in males may be attributed to factors such as hormonal differences, activity levels, and greater susceptibility to trauma. Despite advancements in treatment, approximately 30% of bone infections progress to chronic OM. Chronic OM can be challenging to treat and is associated with ongoing inflammation, bone destruction, and the potential for recurrent infections. Effective management requires timely intervention, appropriate antimicrobial therapy, and, in some cases, surgical debridement or stabilization to prevent further complications [101]. In adults, OM commonly affects individuals with underlying conditions such as diabetes, those who have experienced trauma, or those who have undergone orthopedic surgery. Diabetes increases the risk of OM due to impaired immune response, poor circulation, and the presence of neuropathy, which can lead to unnoticed injuries and infections. Trauma, such as fractures or open wounds, can directly introduce pathogens into the bone. At the same time, orthopedic surgeries, particularly joint replacements or internal fixation procedures, may provide a portal for bacteria to enter the bone, leading to infection. In these cases, timely diagnosis and targeted antimicrobial therapy are critical to prevent the progression to chronic OM, which can lead to significant morbidity [102]. Hip and knee replacement surgeries are among the most common causes of OM in adults. One of the major challenges in treating OM following these procedures is the formation of bacterial biofilms on the implanted foreign materials, such as prosthetic joints. Bacterial biofilms are clusters of bacteria encased in a protective matrix that adhere to surfaces, making them much more resistant to both the immune system and antibiotic treatments. This biofilm formation significantly complicates the treatment of infections, as the bacteria within the biofilm are less susceptible to standard antimicrobial therapies. As a result, surgical intervention, often including the removal of the infected implant, may be necessary in cases of chronic infection. Therefore, preventing biofilm formation and developing strategies to disrupt existing biofilms are key areas of focus in improving outcomes for patients with prosthetic-related OM [103].
Once the bone is infected, polymorphonuclear leukocytes (PMNs), also known as neutrophils, are recruited to the site of infection as part of the body’s innate immune response. These cells attempt to eliminate the infectious organisms by phagocytosing them. However, to effectively destroy the pathogens, activated PMNs release several highly reactive oxidants, including reactive oxygen species (ROS) and reactive nitrogen species (RNS), through processes such as the respiratory burst. While these oxidants play a critical role in pathogen destruction, they can also inadvertently cause damage to surrounding tissue. In the context of OM, the release of these reactive molecules can lead to the destruction of bone tissue, contributing to bone lysis and further exacerbating the infection. This immune-mediated damage to bone is a key factor in the progression of OM, as the bone’s integrity is compromised, and the healing process is impaired. Therefore, understanding and controlling the inflammatory response, including the activity of PMNs and the regulation of oxidative stress (OS), is essential in managing OM and preventing further tissue damage [104]. As the infection progresses, the pus produced by the pathogenic organisms spreads into the blood vessels within the bone, leading to significant alterations in blood flow. This disruption causes localized ischemia and the formation of devitalized, or necrotic, areas of bone. The lack of adequate blood supply to the affected bone tissue results in avascular necrosis, which is a hallmark of OM. In response to the infection and bone death, the body attempts to repair the damage by creating new bone around the necrotic area, a process known as involucrum formation. The involucrum is a layer of new bone that surrounds the dead bone, effectively encapsulating the sequestrum, or the dead bone, which becomes a reservoir for persistent infection. The presence of sequestrum contributes to the chronic nature of the infection, as it can serve as a nidus for ongoing bacterial activity, making it difficult for antibiotics and the immune system to eradicate the infection. This cycle of bone destruction, necrosis, and the body’s attempt at repair often leads to chronic OM, where infection and bone damage persist over time. Surgical removal of the sequestrum and proper antibiotic treatment are crucial for treating chronic OM and preventing further complications [104].
Chronic OM is often caused by persistent bacterial infection, with S. aureus being the most common pathogen responsible. This bacterium is particularly adept at evading the immune system and resisting treatment, which contributes to the persistence of the infection. The ability of S. aureus to form biofilms on bone and foreign materials (such as orthopedic implants) complicates treatment, as biofilms protect the bacteria from both the immune response and antibiotics. In chronic OM, S. aureus can establish a long-lasting infection by forming biofilms around areas of necrotic bone (sequestrum), making it difficult for antibiotics to penetrate and effectively eliminate the bacteria. The biofilm also creates a chronic inflammatory environment, where immune cells such as PMNs are activated but often unable to fully clear the infection. As a result, the infection persists, leading to ongoing bone destruction and the formation of new bone around the infected area (involucrum), which further complicates treatment and the healing process. Effective management of chronic OM requires a combination of surgical intervention to remove necrotic bone and infected tissue, along with prolonged antibiotic therapy that can penetrate biofilms and target the bacteria more effectively [100]. Although S. aureus has remained the most common cause of OM, other pathogens can also be involved, and the specific pathogens often vary with age, underlying conditions, and the route of infection. In children, Escherichia coli is a common causative organism of hematogenous OM, especially in cases associated with urinary tract infections or other systemic conditions. Other gram-negative bacteria, such as Klebsiella spp., Enterobacter spp., and Pseudomonas spp., are more frequently implicated in adults, particularly those with chronic medical conditions, such as diabetes, immunocompromised states, or post-surgical infections. Fungal, viral, and parasitic infections are far less common causes of OM. Fungal infections are typically seen in immunocompromised patients, while viral and parasitic infections causing OM are rare and generally secondary to other systemic infections. Anatomically, hematogenous OM tends to affect bones with ample blood supply, which can lead to more pronounced and rapid dissemination of pathogens. Commonly affected bones include the tibia, femur, humerus, vertebrae, and jaw. These bones are highly vascularized, making them more susceptible to bacterial seeding through the bloodstream during the acute phase of infection [3, 105].
S. aureus and osteoblasts
S. aureus can interact with osteoblasts not only on the cell surface but also intracellularly, due to its capability for internalization. This capability allows the bacteria to evade immune responses and persist within osteoblasts, potentially contributing to chronic infection and bone damage [106]. S. aureus contains various surface components, including cell wall peptidoglycans, lipoteichoic acid, and lipoproteins, which are collectively referred to as pathogen-associated molecular patterns (PAMPs). These PAMPs interact with osteoblasts, triggering the release of chemokines (such as CXCL2, CXCL8, CXCL10, CCL2, CCL3, and CCL5) and cytokines (including IL-1β, IL-18, and TNF-α). These molecules play a crucial role in recruiting and activating both innate immune cells (such as neutrophils and monocytes/macrophages) and adaptive immune cells (like lymphocytes), thereby enhancing the inflammatory response and promoting immune defense against the infection [107, 108]. S. aureus can interfere with bone formation by decreasing the expression of vital osteoblast markers necessary for their growth and proliferation. These markers include alkaline phosphatase, collagen type I, osteopontin, and osteocalcin, which are crucial for osteoblast function and bone matrix production. In addition, S. aureus stimulates osteoblasts to secrete soluble RANKL. This molecule plays a pivotal role in recruiting and activating osteoclasts, the cells responsible for bone resorption. As a result, the balance between bone formation and resorption is disrupted, leading to excessive bone loss and the promotion of inflammation-driven resorption, which contributes to skeletal damage. This mechanism highlights how S. aureus can not only impair osteoblast function but also drive osteoclast-mediated bone destruction, further complicating the inflammatory response in bone infections, such as OM [109, 110].
Ogawa was the first to present evidence showing that osteoblasts can internalize S. aureus [111]. The primary mechanism involves the interaction between fibronectin-binding proteins A and B (Fnbp A/B) on S. aureus and fibronectin, which acts as a mediator by connecting S. aureus to osteoblasts through the α5β1 integrin. This interaction facilitates the adhesion of the bacteria to osteoblasts, promoting infection and potentially influencing the immune response and bone remodeling processes [112].
By entering osteoblasts, S. aureus avoids detection by the immune system, promoting the persistence and spread of the infection. Inside the osteoblast, S. aureus resides within endolysosomal vesicles, where it can survive for prolonged periods [113-115].
S. aureus can persist for extended periods within osteoblasts, as evidenced by the presence of small colony variants. These variants exhibit an atypical, characteristic morphology that reduces their aggressiveness toward the host cell, thereby enhancing their long-term survival. However, despite these prolonged survival rates, the osteoblast’s safety is not guaranteed. Over time, the highly detrimental effects of PSMs on the cell membrane prove lethal, ultimately leading to the osteoblast’s death [115].
In response to S. aureus infection, osteoblasts do not remain inactive; rather, they actively release a variety of inflammatory mediators. These factors are part of the innate immune response and are crucial in initiating the body’s defense against the bacterial pathogen. Simultaneously, this response works in tandem with the adaptive immune system, particularly through the activation of Th1 lymphocytes, which further enhance the immune response and help coordinate the body’s efforts to combat the infection [116].
As previously mentioned, the conflict between S. aureus and osteoblasts is typically prolonged, with S. aureus emerging victorious, leading to subsequent bone loss. S. aureus is capable of inhibiting osteoblast activity and differentiation, while also preventing mineralization [117-120].
S. aureus can also induce osteoblast apoptosis through the activation of multiple pathways, all of which ultimately lead to the same outcome [121-123]. Undoubtedly, the death of osteoblasts is a hallmark event in OM caused by S. aureus, with dual consequences [124].
S. aureus and osteoclasts
Bone loss in OM occurs due to various factors and is not solely caused by the reduced function or death of osteoblasts. A key contributor to this process is the substantial increase in osteoclast differentiation and activity. Osteoblasts regulate osteoclastogenesis through the production of two important proteins: RANKL and OPG. RANKL, when bound to the RANK receptor on osteoclast precursors, triggers their differentiation into mature osteoclasts, which are responsible for bone resorption. On the other hand, OPG, which is produced by osteoblasts in a soluble form, acts as a decoy receptor. It binds to RANKL, preventing its interaction with RANK, and thus controls the differentiation and activation of osteoclasts. This balance between RANKL and OPG is crucial for bone homeostasis, and its disruption during OM leads to excessive osteoclast activity, contributing to bone destruction [125].
During S. aureus infection, osteoblasts elevate the production of RANKL while reducing the production of OPG, thereby enhancing osteoclastogenesis and accelerating bone resorption [110]. Research has also shown that osteoclasts infected with S. aureus produce substantial quantities of prostaglandin E2 (PGE2). This molecule binds to the EP4 receptor on osteoclasts, increasing RANKL production. The interaction between PGE2 and EP4 triggers a marked increase in RANKL levels, which in turn accelerates osteoclastogenesis, enhancing the differentiation and activation of osteoclasts. This mechanism highlights the role of PGE2 as a key mediator in the inflammatory process and bone resorption during S. aureus infection, further contributing to the pathological changes observed in OM [126, 127].
S. aureus (and or its components) can stimulate the release of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β. These cytokines play a key role in osteoclastogenesis by facilitating the differentiation and activation of osteoclasts from preosteoclasts [128, 129].
In animal models of induced OM, as well as in human bone samples and plasma from patients, these cytokines are found to be significantly elevated both in tissue and the circulatory system [130-132]. Osteoblasts secrete IL-6, as well as other mediators like CCL2 (monocyte chemotactic protein-1, MCP-1), CCL3 (macrophage inflammatory protein-1 alpha, MIP-1α), and CXCL-2 (MIP-2), in response to stimulation by S. aureus [133].
It is hypothesized that osteomacs, the macrophages present in bone, may act as additional contributors to the inflammation observed in OM [133].
A significant study demonstrated that the impact of S. aureus infection on osteoclast precursor cells derived from bone marrow is influenced by the stage of differentiation of these cells. When these precursor cells have already committed to the osteoclast lineage, the presence of RANKL promotes their differentiation into active osteoclasts, which are responsible for bone resorption. On the other hand, when the cells are still undifferentiated, RANKL cannot effectively trigger osteoclast differentiation. Instead, this scenario leads to the activation of M1 macrophages, which are known to contribute to the inflammatory process. This distinction highlights the complex relationship between infection, cell differentiation, and bone remodeling, suggesting that the stage of differentiation of osteoclast precursors plays a critical role in the inflammatory and bone-resorptive processes seen during S. aureus infection [9, 134].
S. aureus and osteocytes
Osteoblasts that lose their functional role become entrapped within the bone matrix they create and subsequently differentiate into osteocytes. These osteocytes, which have an irregular shape, extend their cytoplasmic processes into narrow channels known as canaliculi, forming a network that connects them with other osteocytes. This network facilitates the exchange of nutrients and waste products, which is crucial for maintaining the function and survival of osteocytes. Osteocytes play a crucial role in maintaining the integrity of the bone matrix, as they produce enzymes that help preserve the mineralized structure of the bone. Furthermore, osteocytes are capable of selectively removing minerals and restructuring the organic components of the bone, a process known as osteolysis, which plays a role in bone remodeling and maintaining bone health. This explanation highlights the dynamic roles of osteocytes in maintaining bone homeostasis and their active involvement in both bone maintenance and remodeling processes [3].
The involvement of bone remodeling in OM is still being studied. However, research has demonstrated an increase in metalloproteinase expression in osteocytes infected with S. aureus. This finding suggests that S. aureus infection may influence the process of osteolysis, potentially interfering with the overall process of bone remodeling. Osteolysis is crucial for maintaining the balance between bone formation and resorption, and its disruption in the context of OM may contribute to the structural and functional impairments observed in the bone tissue during infection [17].
Research has shown that human osteocyte-like cultures exposed to S. aureus exhibit a significant upregulation in the expression of several chemokines and cytokines, including CXCL9, CXCL10, and CXCL11. This observation suggests that osteocytes play an active role in attracting cytotoxic and or regulatory T-lymphocyte subsets to the sites of infection. By releasing these signaling molecules, osteocytes may contribute to the immune response and further influence the progression of the infection [17, 135].
Role of ROS in bone dynamics
ROS are molecules and free radicals produced as by-products of cellular processes, particularly oxidative phosphorylation in mitochondria. These species play crucial roles in regulating various physiological functions, including cell proliferation, metabolism, differentiation, and apoptosis. However, when their levels become dysregulated, often due to aging, inflammation, or age-related diseases like osteoarthritis, they contribute to OS and cellular damage. ROS also influences bone homeostasis, including the differentiation and activity of osteoclasts. The enzymes responsible for generating ROS, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, are pivotal in the regulation of bone resorption. Understanding the balance of ROS production and its impact on bone cells is crucial for developing therapeutic approaches to bone-related diseases. For a more detailed overview of ROS types and their roles in bone dynamics, refer to Table 2.
ROS signaling in bone remodeling
Bone homeostasis relies on a delicate balance between bone formation and resorption. With aging, this balance is often one of the first to be disrupted, as bone resorption tends to outpace bone formation [154]. ROS are key regulators of bone turnover, and extensive research has explored their role in influencing both bone formation and resorption [155]. Osteosclerosis can interfere with bone remodeling by creating an imbalance that promotes osteoclast activity, leading to the enhanced differentiation of pre-osteoclasts into mature osteoclasts. This disruption in the bone remodeling process may contribute to the development of metabolic bone diseases and skeletal disorders, including osteoporosis [156].
ROS can suppress or even completely halt the activity and differentiation of osteoblasts destined for apoptosis, while also affecting osteocytes. This disruption in osteoblast and osteocyte function further promotes osteoclastogenesis, leading to an imbalance in bone remodeling. ROS-induced alterations in osteoblast and osteocyte behavior can exacerbate the processes of bone resorption, contributing to conditions such as osteoporosis and other metabolic bone diseases [157-159].
Various factors, primarily secreted by osteoblasts and osteocytes, regulate the activity of osteoclasts and osteoblasts, thus influencing bone remodeling. Key factors are RANKL and OPG, as previously discussed. Their expression is closely influenced by an elevated oxidative state, resulting in the upregulation of RANKL and the downregulation of OPG. This process is mediated by the activation of protein kinases (such as ERK1/2, JNK) and/or other factors that modulate specific transcription factors [160-162].
OS inhibits osteoblast activation and reduces the production of OPG, thereby amplifying the effects of RANKL and stimulating osteoclast differentiation and activity. This disruption increases the RANKL/OPG ratio, a critical marker reflecting the degree of bone resorption [163]. An increase in the RANKL/OPG ratio indicates an imbalance that favors bone resorption over bone formation. This disruption is closely associated with the onset of various skeletal diseases, including different forms of osteoporosis and bone disorders secondary to inflammation, such as OM. Under normal conditions, the equilibrium between RANKL and OPG is tightly regulated to ensure a balanced process of bone formation and resorption. However, when this balance is disturbed and the RANKL/OPG ratio increases, bone resorption becomes dominant. This imbalance contributes to the degradation and thinning of bone tissue, which plays a central role in the development of diseases such as osteoporosis and OM. These conditions, often accompanied by inflammation, disrupt the physiological processes of bone remodeling, leading to weakened bones and an increased risk of fractures [3, 162, 164].
A self-perpetuating loop is created between OS and RANKL, where the excessive production of ROS inhibits osteoblast activation and OPG production. This condition increases RANKL, leading to enhanced osteoclastogenesis and subsequent bone resorption. RANKL, in turn, further promotes ROS production through the involvement of various intracellular signaling molecules such as TRAF6, Rac1, and NOX, amplifying the cycle and exacerbating bone degradation [165-167]. Furthermore, given the critical role of NOX in ROS production during osteoclast differentiation, the signaling cascade leading from RANK to ROS production can be outlined as follows: RANK activates TRAF6, which in turn activates Rac1. Rac1 then stimulates the NOX complex, resulting in the production of ROS. This pathway highlights the essential molecular interactions that contribute to ROS generation during osteoclastogenesis [168].
OS biomarkers in OM
Oxidative stress plays a crucial role in the pathophysiology of OM, a severe musculoskeletal infection. OS arises from an imbalance between ROS and the body’s antioxidant defenses, contributing to tissue damage and the progression of disease. Multiple studies have highlighted the potential of OS biomarkers in diagnosing and monitoring OM. For example, paraoxonase-1 activity, serum lipid hydroperoxides, and 8-hydroxy-2-deoxyguanosine (8-OHdG) levels have been shown to correlate with the severity of oxidative damage in OM patients. These markers offer insight into disease status and may inform therapeutic strategies aimed at mitigating oxidative damage and enhancing patient outcomes. For detailed findings on oxidative stress biomarkers in OM, refer to Table 3.
Conclusion
OM represents a significant challenge in both clinical and molecular research due to its complex pathophysiology and impact on bone health. The molecular mechanisms underlying the infection involve a coordinated response between the immune system and bone cells, resulting in inflammation, bone resorption, and impaired bone healing. Understanding these molecular pathways is essential for identifying novel therapeutic targets that could enhance the treatment of OM. Further research is needed to explore the cellular interactions involved and to develop more effective strategies for preventing bone destruction and promoting bone regeneration in affected individuals.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Conceptualization and supervision: Abolfazl Bagherifard and Hooman Yahyazedeh; Validation : Hooman Yahyazedeh and Khatere Mokhtari; Writing the original draft: Khatere Mokhtari and Abolfazl Bagherifard; Investigation, review and editing: All authors.
Conflict of interest
The authors declared no conflict of interest.
References
- Xing L, Schwarz EM, Boyce BF. Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev. 2005; 208:19-29. [DOI:10.1111/j.0105-2896.2005.00336.x] [PMID]
- Zhao Q, Shao J, Chen W, Li YP. Osteoclast differentiation and gene regulation. Front Biosci. 2007; 12:2519-29.[DOI:10.2741/2252] [PMID]
- Massaccesi L, Galliera E, Pellegrini A, Banfi G, Corsi Romanelli MM. Osteomyelitis, oxidative stress and related biomarkers. Antioxidants (Basel). 2022; 11(6):1061. [DOI:10.3390/antiox11061061]
- Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol. 2008; 3(Suppl 3):S131-9. [DOI:10.2215/CJN.04151206] [PMID]
- Baniasadi M, Talebi S, Mokhtari K, Zabolian AH, Khosroshahi EM, Entezari M, et al. Role of non-coding RNAs in osteoporosis. Pathol Res Pract. 2024; 253:155036. [DOI:10.1016/j.prp.2023.155036] [PMID]
- Amroodi MN, Maghsoudloo M, Amiri S, Mokhtari K, Mohseni P, Pourmarjani A, et al. Unraveling the molecular and immunological landscape: Exploring signaling pathways in osteoporosis. Biomed Pharmacother. 2024; 177:116954. [DOI:10.1016/j.biopha.2024.116954]
- Manolagas SC. Steroids and osteoporosis: The quest for mechanisms. J Clin Invest. 2013; 123(5):1919-21. [DOI:10.1172/JCI68062] [PMID]
- Mbalaviele G, Novack DV, Schett G, Teitelbaum SL. Inflammatory osteolysis: A conspiracy against bone. J Clin Invest. 2017; 127(6):2030-9. [DOI:10.1172/JCI93356]
- Trouillet-Assant S, Gallet M, Nauroy P, Rasigade JP, Flammier S, Parroche P, et al. Dual impact of live Staphylococcus aureus on the osteoclast lineage, leading to increased bone resorption. J Infect Dis. 2015; 211(4):571-81. [DOI:10.1093/infdis/jiu386] [PMID]
- Chen Y, Liu Z, Lin Z, Lu M, Fu Y, Liu G, et al. The effect of Staphylococcus aureus on innate and adaptive immunity and potential immunotherapy for S. aureus-induced osteomyelitis. Front Immunol. 2023; 14:1219895. [DOI:10.3389/fimmu.2023.1219895] [PMID]
- Guerra FE, Borgogna TR, Patel DM, Sward EW, Voyich JM. Epic immune battles of history: Neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol. 2017; 7:286. [DOI:10.3389/fcimb.2017.00286] [PMID]
- Gimza BD, Cassat JE. Mechanisms of antibiotic failure during staphylococcus aureus osteomyelitis. Front Immunol. 2021; 12:638085. [DOI:10.3389/fimmu.2021.638085] [PMID]
- Stoodley P, Nistico L, Johnson S, Lasko LA, Baratz M, Gahlot V, et al. Direct demonstration of viable Staphylococcus aureus biofilms in an infected total joint arthroplasty. A case report. J Bone Joint Surg Am. 2008; 90(8):1751-8. [DOI:10.2106/JBJS.G.00838] [PMID]
- Horn CM, Kielian T. Crosstalk between staphylococcus aureus and innate immunity: Focus on immunometabolism. Front Immunol. 2021; 11:621750. [DOI:10.3389/fimmu.2020.621750] [PMID]
- Tucey TM, Verma J, Harrison PF, Snelgrove SL, Lo TL, Scherer AK, et al. Glucose homeostasis is important for immune cell viability during candida challenge and host survival of systemic fungal infection. Cell Metab. 2018; 27(5):988-1006.e7. [DOI:10.1016/j.cmet.2018.03.019]
- Flannagan RS, Heit B, Heinrichs DE. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol. 2016; 18(4):514-35. [DOI:10.1111/cmi.12527] [PMID]
- Yang D, Wijenayaka AR, Solomon LB, Pederson SM, Findlay DM, Kidd SP, et al. Novel insights into staphylococcus aureus deep bone infections: The Involvement of osteocytes. mBio. 2018; 9(2):e00415-18. [DOI:10.1128/mBio.00415-18]
- Alder KD, Lee I, Munger AM, Kwon HK, Morris MT, Cahill SV, et al. Intracellular Staphylococcus aureus in bone and joint infections: A mechanism of disease recurrence, inflammation, and bone and cartilage destruction. Bone. 2020; 141:115568. [DOI:10.1016/j.bone.2020.115568]
- Krishna S, Miller LS. Innate and adaptive immune responses against Staphylococcus aureus skin infections. Semin Immunopathol. 2012; 34(2):261-80. [DOI: 10.1007/s00281-011-0292-6] [PMID]
- de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med. 2004; 199(5):687-95. [DOI:10.1084/jem.20031636] [PMID]
- Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*). Annu Rev Immunol. 2009; 27:551-89. [DOI:10.1146/annurev.immunol.021908.132723] [PMID]
- Bardoel BW, Vos R, Bouman T, Aerts PC, Bestebroer J, Huizinga EG, et al. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J Mol Med (Berl). 2012; 90(10):1109-20. [DOI:10.1007/s00109-012-0926-8] [PMID]
- Yokoyama R, Itoh S, Kamoshida G, Takii T, Fujii S, Tsuji T, et al. Staphylococcal superantigen-like protein 3 binds to the Toll-like receptor 2 extracellular domain and inhibits cytokine production induced by Staphylococcus aureus, cell wall component, or lipopeptides in murine macrophages. Infect Immun. 2012; 80(8):2816-25. [DOI:10.1128/IAI.00399-12]
- Stapels DA, Ramyar KX, Bischoff M, von Köckritz-Blickwede M, Milder FJ, Ruyken M, et al. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc Natl Acad Sci U S A. 2014; 111(36):13187-92. [DOI:10.1073/pnas.1407616111] [PMID]
- Wagner C, Iking-Konert C, Hug F, Stegmaier S, Heppert V, Wentzensen A, et al. Cellular inflammatory response to persistent localized Staphylococcus aureus infection: Phenotypical and functional characterization of polymorphonuclear neutrophils (PMN). Clin Exp Immunol. 2006; 143(1):70-7. [DOI:10.1111/j.1365-2249.2005.02963.x] [PMID]
- Seebach E, Kubatzky KF. Chronic implant-related bone infections-can immune modulation be a therapeutic strategy? Front Immunol. 2019; 10:1724. [DOI:10.3389/fimmu.2019.01724] [PMID]
- Günther F, Wabnitz GH, Stroh P, Prior B, Obst U, Samstag Y, et al. Host defence against Staphylococcus aureus biofilms infection: Phagocytosis of biofilms by polymorphonuclear neutrophils (PMN). Mol Immunol. 2009; 46(8-9):1805-13. [DOI:10.1016/j.molimm.2009.01.020] [PMID]
- Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology (Reading). 2003; 149(Pt 10):2749-58. [DOI:10.1099/mic.0.26353-0]
- Cosgrove K, Coutts G, Jonsson IM, Tarkowski A, Kokai-Kun JF, Mond JJ, et al. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol. 2007; 189(3):1025-35. [DOI:10.1128/JB.01524-06] [PMID]
- Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, et al. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005; 202(2):209-15. [DOI:10.1084/jem.20050846] [PMID]
- Vozza EG, Mulcahy ME, McLoughlin RM. Making the most of the host; Targeting the autophagy pathway facilitates Staphylococcus aureus intracellular survival in neutrophils. Front Immunol. 2021; 12:667387. [DOI:10.3389/fimmu.2021.667387]
- Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A. Neutrophil extracellular traps: How to generate and visualize them. J Vis Exp. 2010; (36):1724. [DOI:10.3791/1724-v] [PMID]
- Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood. 2011; 118(7):1952-61. [DOI:10.1182/blood-2011-03-343061] [PMID]
- Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity. 2010; 32(5):593-604. [DOI:10.1016/j.immuni.2010.05.007] [PMID]
- Wang Y, Lin Y, Cheng C, Chen P, Zhang P, Wu H, et al. NF-κB/TWIST1 mediates migration and phagocytosis of macrophages in the mice model of implant-associated staphylococcus aureus osteomyelitis. Front Microbiol. 2020; 11:1301 [DOI:10.3389/fmicb.2020.01301]
- Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, et al. Macrophage polarization in chronic inflammatory diseases: Killers or builders? J Immunol Res. 2018; 2018:8917804.[DOI:10.1155/2018/8917804] [PMID]
- Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest. 2012; 122(3):787-95. [DOI:10.1172/JCI59643]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002; 169(5):2253-63. [DOI:10.4049/jimmunol.169.5.2253] [PMID]
- Cheng CI, Chen PH, Lin YC, Kao YH. High glucose activates Raw264. 7 macrophages through RhoA kinase-mediated signaling pathway. Cell Signal. 2015; 27(2):283-92. [DOI:10.1016/j.cellsig.2014.11.012] [PMID]
- Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018; 233(9):6425-40. [DOI:10.1002/jcp.26429] [PMID]
- Ding X, Li F, Zhang L. Knockdown of Delta-like 3 restricts lipopolysaccharide-induced inflammation, migration and invasion of A2058 melanoma cells via blocking Twist1-mediated epithelial-mesenchymal transition. Life Sci. 2019; 226:149155. [DOI:10.1016/j.lfs.2019.04.024]
- Lv Y, Fang L, Ding P, Liu R. PI3K/Akt-Beclin1 signaling pathway positively regulates phagocytosis and negatively mediates NF-κB-dependent inflammation in Staphylococcus aureus-infected macrophages. Biochem Biophys Res Commun. 2019; 510(2):284-9. [DOI:10.1016/j.bbrc.2019.01.091] [PMID]
- Berends ETM, Zheng X, Zwack EE, Ménager MM, Cammer M, Shopsin B, et al. Staphylococcus aureus impairs the function of and kills human dendritic cells via the LukAB Toxin. mBio. 2019; 10(1):e01918-18. [DOI:10.1128/mBio.01918-18] [PMID]
- Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U S A. 2021; 118(30):e2023739118. [DOI:10.1073/pnas.2023739118]
- Seo W, Jerin C, Nishikawa H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp Mol Med. 2021; 53(2):202-9. [DOI:10.1038/s12276-021-00568-0] [PMID]
- Kurachi M. CD8+ T cell exhaustion. Semin immunopathol. 2019; 41(3):327-37. [DOI: 10.1007/s00281-019-00744-5] [PMID]
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity. 2016; 45(2):358-73. [DOI:10.1016/j.immuni.2016.07.008]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009; 10(1):29-37. [DOI:10.1038/ni.1679]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011; 35(6):871-82. [DOI:10.1016/j.immuni.2011.09.021] [PMID]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439(7077):682-7. [DOI:10.1038/nature04444] [PMID]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005; 25(21):9543-53. [DOI:10.1128/MCB.25.21.9543-9553.2005] [PMID]
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015; 6:6692. [DOI:10.1038/ncomms7692]
- Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T Cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017; 32(3):377-91.e9. [DOI:10.1016/j.ccell.2017.08.004] [PMID]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005; 6(12):1236-44. [DOI:10.1038/ni1268] [PMID]
- Li K, Chen Y, Lin Y, Zhang G, Su J, Wu X, et al. PD-1/PD-L1 blockade is a potent adjuvant in treatment of Staphylococcus aureus osteomyelitis in mice. Mol Ther. 2023 ; 31(1):174-92.[DOI:10.1016/j.ymthe.2022.09.006]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016; 537(7620):412-28. [DOI:10.1038/nature19317]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity. 2016; 45(2):415-27. [DOI:10.1016/j.immuni.2016.07.021] [PMID]
- Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity. 2019; 51(5):840-55.e5. [DOI: 10.1016/j.immuni.2019.09.013] [PMID]
- Diller ML, Kudchadkar RR, Delman KA, Lawson DH, Ford ML. Balancing Inflammation: The link between Th17 and Regulatory T Cells. Mediators Inflamm. 2016; 2016:6309219. [DOI:10.1155/2016/6309219] [PMID]
- Wang M, Tian T, Yu S, He N, Ma D. Th17 and Treg cells in bone related diseases. Clin Dev Immunol. 2013; 2013:203705. [DOI:10.1155/2013/203705] [PMID]
- Tyagi AM, Yu M, Darby TM, Vaccaro C, Li JY, Owens JA, et al., The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of WNT10B expression. Immunity. 2018; 49(6):1116-31.e7. e7. [DOI:10.1016/j.immuni.2018.10.013]
- Björkander S, Hell L, Johansson MA, Forsberg MM, Lasaviciute G, Roos S, et al. Staphylococcus aureus-derived factors induce IL-10, IFN-γ and IL-17A-expressing FOXP3+CD161+ T-helper cells in a partly monocyte-dependent manner. Sci Rep. 2016; 6:22083. [DOI:10.1038/srep22083] [PMID]
- Thomsen IP. Antibody-based intervention against the pore-forming toxins of Staphylococcus aureus. Virulence. 2018; 9(1):645-7. [DOI:10.1080/21505594.2017.1421831] [PMID]
- Thomsen IP, Sapparapu G, James DBA, Cassat JE, Nagarsheth M, Kose N, et al. Monoclonal antibodies against the staphylococcus aureus bicomponent leukotoxin AB isolated following invasive human infection reveal diverse binding and modes of action. J Infect Dis. 2017; 215(7):1124-31. [DOI:10.1093/infdis/jix071]
- Goldmann O, Medina E. Staphylococcus aureus strategies to evade the host acquired immune response. Int J Med Microbiol. 2018; 308(6):625-30. [DOI:10.1016/j.ijmm.2017.09.013] [PMID]
- Goodyear CS, Silverman GJ. Death by a B cell superantigen: In vivo VH-targeted apoptotic supraclonal B cell deletion by a Staphylococcal Toxin. J Exp Med. 2003; 197(9):1125-39. [DOI:10.1084/jem.20020552] [PMID]
- Becker S, Frankel MB, Schneewind O, Missiakas D. Release of protein A from the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2014; 111(4):1574-9. [DOI:10.1073/pnas.1317181111] [PMID]
- Rooijakkers SH, van Wamel WJ, Ruyken M, van Kessel KP, van Strijp JA. Anti-opsonic properties of staphylokinase. Microbes Infect. 2005; 7(3):476-84. [DOI:10.1016/j.micinf.2004.12.014]
- Pelzek AJ, Shopsin B, Radke EE, Tam K, Ueberheide BM, Fenyö D, et al. Human memory B cells targeting Staphylococcus aureus exotoxins are prevalent with skin and soft tissue infection. mBio. 2018; 9(2):e02125-17. [DOI:10.1128/mBio.02125-17]
- Kang SS, Kim SK, Baik JE, Ko EB, Ahn KB, Yun CH, et al. Staphylococcal LTA antagonizes the B cell-mitogenic potential of LPS. Sci Rep. 2018; 8(1):1496. [DOI:10.1038/s41598-018-19653-y] [PMID]
- Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006; 169(3):987-98. [DOI:10.2353/ajpath.2006.060180] [PMID]
- Liu ZQ, Wu Y, Song JP, Liu X, Liu Z, Zheng PY, et al. Tolerogenic CX3CR1+ B cells suppress food allergy-induced intestinal inflammation in mice. Allergy. 2013; 68(10):1241-8. [DOI:10.1111/all.12218] [PMID]
- Nouël A, Pochard P, Simon Q, Ségalen I, Le Meur Y, Pers JO, et al. B-Cells induce regulatory T cells through TGF-β/IDO production in A CTLA-4 dependent manner. J Autoimmun. 2015; 59:53-60. [DOI:10.1016/j.jaut.2015.02.004]
- Wu H, Jia C, Wang X, Shen J, Tan J, Wei Z, et al. The impact of methicillin resistance on clinical outcome among patients with Staphylococcus aureus osteomyelitis: A retrospective cohort study of 482 cases. Sci Rep. 2023; 13(1):7990. [DOI:10.1038/s41598-023-35111-w] [PMID]
- Lee CC, Southgate RD, Jiao C, Gersz E, Owen JR, Kates SL, et al. Deriving a dose and regimen for anti-glucosaminidase antibody passive-immunisation for patients with Staphylococcus aureus osteomyelitis. Eur Cell Mater. 2020; 39:96-107. [DOI:10.22203/eCM.v039a06]
- McCulloch TR, Wells TJ, Souza-Fonseca-Guimaraes F. Towards efficient immunotherapy for bacterial infection. Trends Microbiol. 2022; 30(2):158-69. [DOI:10.1016/j.tim.2021.05.005]
- Otto M. Novel targeted immunotherapy approaches for staphylococcal infection. Expert Opin Biol Ther. 2010; 10(7):1049-59. [DOI:10.1517/14712598.2010.495115] [PMID]
- Harro JM, Achermann Y, Freiberg JA, Allison DL, Brao KJ, Marinos DP, et al. Clearance of Staphylococcus aureus from In vivo models of chronic infection by immunization requires both planktonic and biofilm antigens. Infect Immun. 2019; 88(1):e00586-19. [PMID]
- Codarri Deak L, Hashimoto M, Umaña P, Klein C. Beyond checkpoint inhibition: PD-1 cis-targeting of an IL-2Rβγ-biased interleukin-2 variant as a novel approach to build on checkpoint inhibition. Oncoimmunology. 2023; 12(1):2197360. [DOI:10.1080/2162402X.2023.2197360]
- Mao QF, Shang-Guan ZF, Chen HL, Huang K. Immunoregulatory role of IL-2/STAT5/CD4+CD25+Foxp3 Treg pathway in the pathogenesis of chronic osteomyelitis. Ann Transl Med. 2019; 7(16):384. [DOI:10.21037/atm.2019.07.45][PMID]
- Luo J, Yang J, Peng M, Liu F, Zhou X, Yin H, et al. Efficacy and safety of Chinese herbal medicine Wuwei Xiaodu Drink for wound infection: A protocol for systematic review and meta-analysis. Medicine (Baltimore). 2022; 101(48):e32135.[DOI:10.1097/MD.0000000000032135] [PMID]
- Huang K, Ren HY, Lin BY, Liu YY, Guo QF. Protective effects of Wuwei Xiaodu Drink against chronic osteomyelitis through Foxp3+CD25+CD4+ Treg cells via the IL-2/STAT5 signaling pathway. Chin J Nat Med. 2022; 20(3):185-93.[DOI:10.1016/S1875-5364(22)60146-8]
- Kim JH, Kim EY, Lee B, Min JH, Song DU, Lim JM, et al. The effects of lycii radicis cortex on RANKL-induced osteoclast differentiation and activation in RAW 264.7 cells. Int J Mol Med. 2016; 37(3):649-58. [DOI:10.3892/ijmm.2016.2477]
- Kitaura H, Kimura K, Ishida M, Kohara H, Yoshimatsu M, Takano-Yamamoto T. Immunological reaction in TNF-α-mediated osteoclast formation and bone resorption in vitro and in vivo. Clin Dev Immunol. 2013; 2013:181849. [DOI:10.1155/2013/181849] [PMID]
- Wu Q, Zhou X, Huang D, Ji Y, Kang F. IL-6 Enhances osteocyte-mediated osteoclastogenesis by promoting JAK2 and RANKL activity in vitro. Cell Physiol Biochem. 2017; 41(4):1360-9. [DOI:10.1159/000465455]
- Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL. M-CSF mediates TNF-induced inflammatory osteolysis. J Clin Invest. 2005; 115(12):3418-27. [DOI:10.1172/JCI26132] [PMID]
- Kanazawa K, Kudo A. TRAF2 is essential for TNF-alpha-induced osteoclastogenesis. J Bone Miner Res. 2005; 20(5):840-7. [DOI:10.1359/JBMR.041225]
- Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. 2005; 202(5):589-95.[DOI:10.1084/jem.20050978] [PMID]
- Li P, Schwarz EM, O'Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res. 2004; 19(2):207-13. [DOI:10.1359/JBMR.0301233]
- Lampiasi N, Russo R, Zito F. The alternative faces of macrophage generate osteoclasts. Biomed Res Int. 2016; 2016:9089610. [DOI:10.1155/2016/9089610] [PMID]
- Li X, Zhang W. [Tumor necrosis factor-α promotes osteoclast differentiation via sialylation in mice (Chinese)]. Nan Fang Yi Ke Da Xue Xue Bao. 2021; 41(12):1773-79. [PMID]
- Zaiss MM, Axmann R, Zwerina J, Polzer K, Gückel E, Skapenko A, et al. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheum. 2007; 56(12):4104-12. [DOI:10.1002/art.23138] [PMID]
- Shashkova EV, Trivedi J, Cline-Smith AB, Ferris C, Buchwald ZS, Gibbs J, et al. Osteoclast-Primed Foxp3+ CD8 T Cells Induce T-bet, Eomesodermin, and IFN-γ To Regulate Bone Resorption. J Immunol. 2016; 197(3):726-35 [DOI:10.4049/jimmunol.1600253]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006; 24(2):179-89. [DOI:10.1016/j.immuni.2006.01.001] [PMID]
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006; 441(7090):231-4. [DOI:10.1038/nature04754]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006; 441(7090):235-8. [PMID]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011; 208(7):1367-76. [DOI: 10.1084/jem.20110278] [PMID]
- Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORγ. Cell Rep. 2012; 1(4):360-73.[DOI:10.1016/j.celrep.2012.02.007] [PMID]
- Muthukrishnan G, Masters EA, Daiss JL, Schwarz EM. Mechanisms of immune evasion and bone tissue colonization that make Staphylococcus aureus the primary pathogen in osteomyelitis. Curr Osteoporos Rep. 2019; 17(6):395-404. [DOI:10.1007/s11914-019-00548-4]
- Pimentel de Araujo F, Monaco M, Del Grosso M, Pirolo M, Visca P, et al. Staphylococcus aureus clones causing osteomyelitis: A literature review (2000-2020). J Glob Antimicrob Resist. 2021; 26:29-36. [DOI:10.1016/j.jgar.2021.03.030] [PMID]
- Roesgen M, Hierholzer G, Hax PM. Post-traumatic osteomyelitis. Pathophysiology and management. Arch Orthop Trauma Surg. 1989; 108(1):1-9. [DOI:10.1007/BF00934149]
- Beck-Broichsitter BE, Smeets R, Heiland M. Current concepts in pathogenesis of acute and chronic osteomyelitis. Curr Opin Infect Dis. 2015; 28(3):240-5. [DOI:10.1097/QCO.0000000000000155] [PMID]
- Mauffrey C, Herbert B, Young H, Wilson ML, Hake M, Stahel PF. The role of biofilm on orthopaedic implants: The "holy grail" of post-traumatic infection management? Eur J Trauma Emerg Surg. 2016; 42(4):411-6. [DOI:10.1007/s00068-016-0694-1]
- Lew DP, Waldvogel FA. Osteomyelitis. Lancet. 2004 Jul 24-3; 364(9431):369-79. [PMID]
- Jaramillo D, Dormans JP, Delgado J, Laor T, St Geme JW 3rd. Hematogenous osteomyelitis in infants and children: imaging of a changing disease. Radiology. 2017; 283(3):629-43 [DOI:10.1148/radiol.2017151929] [PMID]
- Hudson MC, Ramp WK, Nicholson NC, Williams AS, Nousiainen MT. Internalization of Staphylococcus aureus by cultured osteoblasts. Microb Pathog. 1995; 19(6):409-19. [DOI:10.1006/mpat.1995.0075]
- Heilmann C. Adhesion mechanisms of staphylococci. Adv Exp Med Biol. 2011; 715:105-23. [DOI:10.1007/978-94-007-0940-9_7]
- Claro T, Widaa A, O'Seaghdha M, Miajlovic H, Foster TJ, O'Brien FJ, et al. Staphylococcus aureus protein A binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS One. 2011; 6(4):e18748. [PMID]
- Tucker KA, Reilly SS, Leslie CS, Hudson MC. Intracellular Staphylococcus aureus induces apoptosis in mouse osteoblasts. FEMS Microbiol Lett. 2000; 186(2):151-6. [DOI:10.1111/j.1574-6968.2000.tb09096.x]
- Widaa A, Claro T, Foster TJ, O'Brien FJ, Kerrigan SW. Staphylococcus aureus protein A plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS One. 2012; 7(7):e40586. [DOI:10.1371/journal.pone.0040586] [PMID]
- Ogawa SK, Yurberg ER, Hatcher VB, Levitt MA, Lowy FD. Bacterial adherence to human endothelial cells in vitro. Infect Immun. 1985; 50(1):218-24. [DOI:10.1128/iai.50.1.218-224.1985]
- Khalil H, Williams RJ, Stenbeck G, Henderson B, Meghji S, Nair SP. Invasion of bone cells by Staphylococcus epidermidis. Microbes Infect. 2007; 9(4):460-5. [DOI:10.1016/j.micinf.2007.01.002] [PMID]
- Grosz M, Kolter J, Paprotka K, Winkler AC, Schäfer D, Chatterjee SS, et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell Microbiol. 2014; 16(4):451-65. [DOI:10.1111/cmi.12233]
- Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, et al. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe. 2013; 13(6):759-72. [DOI:10.1016/j.chom.2013.05.003]
- Rasigade JP, Trouillet-Assant S, Ferry T, Diep BA, Sapin A, Lhoste Y, et al. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS One. 2013; 8(5):e63176. [DOI:10.1371/journal.pone.0063176] [PMID]
- Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007; 7(4):292-304. [PMID]
- Claro T, Widaa A, McDonnell C, Foster TJ, O'Brien FJ, Kerrigan SW. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology (Reading). 2013; 159(Pt 1):147-54. [DOI:10.1099/mic.0.063016-0]
- Jin T, Zhu YL, Li J, Shi J, He XQ, Ding J, et al. Staphylococcal protein A, Panton-Valentine leukocidin and coagulase aggravate the bone loss and bone destruction in osteomyelitis. Cell Physiol Biochem. 2013; 32(2):322-33.[DOI:10.1159/000354440]
- Sanchez CJ Jr, Ward CL, Romano DR, Hurtgen BJ, Hardy SK, Woodbury RL, et al. Staphylococcus aureus biofilms decrease osteoblast viability, inhibits osteogenic differentiation, and increases bone resorption in vitro. BMC Musculoskelet Disord. 2013; 14:187. [DOI:10.1186/1471-2474-14-187] [PMID]
- Chen Q, Hou T, Luo F, Wu X, Xie Z, Xu J. Involvement of toll-like receptor 2 and pro-apoptotic signaling pathways in bone remodeling in osteomyelitis. Cell Physiol Biochem. 2014; 34(6):1890-900. [PMID]
- Alexander EH, Bento JL, Hughes FM Jr, Marriott I, Hudson MC, Bost KL. Staphylococcus aureus and Salmonella enterica serovar Dublin induce tumor necrosis factor-related apoptosis-inducing ligand expression by normal mouse and human osteoblasts. Infect Immun. 2001; 69(3):1581-6. [DOI:10.1128/IAI.69.3.1581-1586.2001]
- Young AB, Cooley ID, Chauhan VS, Marriott I. Causative agents of osteomyelitis induce death domain-containing TNF-related apoptosis-inducing ligand receptor expression on osteoblasts. Bone. 2011 ; 48(4):857-63. [DOI:10.1016/j.bone.2010.11.015]
- Alexander EH, Rivera FA, Marriott I, Anguita J, Bost KL, Hudson MC. Staphylococcus aureus - induced tumor necrosis factor - related apoptosis - inducing ligand expression mediates apoptosis and caspase-8 activation in infected osteoblasts. BMC Microbiol. 2003; 3:5.[DOI:10.1186/1471-2180-3-5]
- Ellington JK, Harris M, Webb L, Smith B, Smith T, Tan K, Hudson M. Intracellular Staphylococcus aureus. A mechanism for the indolence of osteomyelitis. J Bone Joint Surg Br. 2003; 85(6):918-21. [PMID]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003; 423(6937):337-42. [DOI:10.1038/nature01658] [PMID]
- Somayaji SN, Ritchie S, Sahraei M, Marriott I, Hudson MC. Staphylococcus aureus induces expression of receptor activator of NF-kappaB ligand and prostaglandin E2 in infected murine osteoblasts. Infect Immun. 2008; 76(11):5120-6. [DOI:10.1128/IAI.00228-08]
- Jiang W, Jin Y, Zhang S, Ding Y, Huo K, Yang J, et al. PGE2 activates EP4 in subchondral bone osteoclasts to regulate osteoarthritis. Bone Res. 2022; 10(1):27. [DOI:10.1038/s41413-022-00201-4]
- Redlich K, Smolen JS. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012; 11(3):234-50. [PMID]
- Souza PP, Lerner UH. The role of cytokines in inflammatory bone loss. Immunol Invest. 2013; 42(7):555-622. [DOI:10.3109/08820139.2013.822766] [PMID]
- García-Alvarez F, Navarro-Zorraquino M, Castro A, Grasa JM, Pastor C, et al. Effect of age on cytokine response in an experimental model of osteomyelitis. Biogerontology. 2009; 10(5):649-58. [DOI:10.1007/s10522-008-9211-1]
- Marriott I, Gray DL, Tranguch SL, Fowler VG Jr, Stryjewski M, Scott Levin L, et al. Osteoblasts express the inflammatory cytokine interleukin-6 in a murine model of Staphylococcus aureus osteomyelitis and infected human bone tissue. Am J Pathol. 2004; 164(4):1399-406. [DOI:10.1016/S0002-9440(10)63226-9]
- Yoshii T, Magara S, Miyai D, Nishimura H, Kuroki E, Furudoi S, et al. Local levels of interleukin-1beta, -4, -6 and tumor necrosis factor alpha in an experimental model of murine osteomyelitis due to staphylococcus aureus. Cytokine. 2002; 19(2):59-65. [DOI:10.1006/cyto.2002.1039] [PMID]
- Josse J, Velard F, Gangloff SC. Staphylococcus aureus vs. Osteoblast: Relationship and consequences in osteomyelitis. Front Cell Infect Microbiol. 2015; 5:85. [DOI:10.3389/fcimb.2015.00085] [PMID]
- Bar-Shavit Z. Taking a toll on the bones: regulation of bone metabolism by innate immune regulators. Autoimmunity. 2008; 41(3):195-203. [DOI:10.1080/08916930701694469]
- Groom JR, Luster AD. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011; 89(2):207-15. [DOI:10.1038/icb.2010.158] [PMID]
- Agidigbi TS, Kim C. Reactive oxygen species in osteoclast differentiation and possible pharmaceutical targets of ROS-mediated osteoclast diseases. Int J Mol Sci. 2019; 20(14):3576. [DOI: 10.3390/ijms20143576] [PMID]
- Collins JA, Diekman BO, Loeser RF. Targeting aging for disease modification in osteoarthritis. Curr Opin Rheumatol. 2018; 30(1):101-7. [DOI:10.1097/BOR.0000000000000456]
- Go YM, Jones DP. Redox theory of aging: implications for health and disease. Clin Sci (Lond). 2017; 131(14):1669-88.[DOI:10.1042/CS20160897]
- Altindag O, Erel O, Soran N, Celik H, Selek S. Total oxidative/anti-oxidative status and relation to bone mineral density in osteoporosis. Rheumatol Int. 2008; 28(4):317-21. [DOI:10.1007/s00296-007-0452-0] [PMID]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014; 94(3):909-50. [DOI:10.1152/physrev.00026.2013]
- Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest. 1990; 85(3):632-9. [DOI:10.1172/JCI114485] [PMID]
- Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012; 110(10):1364-90. [DOI:10.1161/CIRCRESAHA.111.243972] [PMID]
- Xu Q, Choksi S, Qu J, Jang J, Choe M, Banfi B, Engelhardt JF, et al. NADPH oxidases are essential for macrophage differentiation. J Biol Chem. 2016; 291(38):20030-41. [DOI:10.1074/jbc.M116.731216] [PMID]
- Kang IS, Kim C. NADPH oxidase gp91phox contributes to RANKL-induced osteoclast differentiation by upregulating NFATc1. Sci Rep. 2016; 6:38014. [DOI:10.1038/srep38014]
- Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005; 106(3):852-9. [DOI:10.1182/blood-2004-09-3662] [PMID]
- Kim JH, Kim K, Kim I, Seong S, Kim N. NRROS Negatively regulates osteoclast differentiation by inhibiting RANKL-Mediated NF-N:B and reactive oxygen species pathways. Mol Cells. 2015; 38(10):904-10. [DOI:10.14348/molcells.2015.0177] [PMID]
- Yang S, Zhang Y, Ries W, Key L. Expression of Nox4 in osteoclasts. J Cell Biochem. 2004; 92(2):238-48. [DOI:10.1002/jcb.20048]
- Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, et al. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest. 2013; 123(11):4731-8. [DOI:10.1172/JCI67603]
- Schröder K. NADPH oxidases in bone homeostasis and osteoporosis. Free Radic Biol Med. 2019; 132:67-72. [DOI:10.1016/j.freeradbiomed.2018.08.036] [PMID]
- Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014; 2:535-62. [DOI:10.1016/j.redox.2014.02.006]
- Sies H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015; 4:180-3. [DOI:10.1016/j.redox.2015.01.002] [PMID]
- Baek KH, Oh KW, Lee WY, Lee SS, Kim MK, Kwon HS, et al. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif Tissue Int. 2010; 87(3):226-35. [DOI:10.1007/s00223-010-9393-9] [PMID]
- Yao H, Yao Z, Zhang S, Zhang W, Zhou W. Upregulation of SIRT1 inhibits H2O2‑induced osteoblast apoptosis via FoxO1/β‑catenin pathway. Mol Med Rep. 2018; 17(5):6681-90. [DOI:10.3892/mmr.2018.8657] [PMID]
- Chen X, Wang Z, Duan N, Zhu G, Schwarz EM, Xie C. Osteoblast-osteoclast interactions. Connect Tissue Res. 2018; 59(2):99-107. [DOI:10.1080/03008207.2017.1290085]
- Ohyama Y, Ito J, Kitano VJ, Shimada J, Hakeda Y. The polymethoxy flavonoid sudachitin suppresses inflammatory bone destruction by directly inhibiting osteoclastogenesis due to reduced ROS production and MAPK activation in osteoclast precursors. PLoS One. 2018 ;13(1):e0191192.[DOI:10.1371/journal.pone.0191192] [PMID]
- Banfi G, Iorio EL, Corsi MM. Oxidative stress, free radicals and bone remodeling. Clin Chem Lab Med. 2008; 46(11):1550-5. [DOI:10.1515/CCLM.2008.302] [PMID]
- Romagnoli C, Marcucci G, Favilli F, Zonefrati R, Mavilia C, Galli G, et al. Role of GSH/GSSG redox couple in osteogenic activity and osteoclastogenic markers of human osteoblast-like SaOS-2 cells. FEBS J. 2013; 280(3):867-79. [DOI:10.1111/febs.12075] [PMID]
- Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. 2013; 54(2):264-71. [DOI:10.1016/j.bone.2012.11.038] [PMID]
- Bai XC, Lu D, Bai J, Zheng H, Ke ZY, Li XM, et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem Biophys Res Commun. 2004; 314(1):197-207. [DOI:10.1016/j.bbrc.2003.12.073] [PMID]
- Fontani F, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: Involvement of JNK and ERK1/2 signalling. Calcif Tissue Int. 2015; 96(4):335-46. [DOI:10.1007/s00223-015-9961-0]
- Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014; 94(1):25-34. [DOI:10.1007/s00223-013-9774-y] [PMID]
- Mulcahy LE, Taylor D, Lee TC, Duffy GP. RANKL and OPG activity is regulated by injury size in networks of osteocyte-like cells. Bone. 2011; 48(2):182-8. [DOI:10.1016/j.bone.2010.09.014]
- Lean JM, Jagger CJ, Kirstein B, Fuller K, Chambers TJ. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology. 2005; 146(2):728-35. [DOI:10.1210/en.2004-1021] [PMID]
- Lang S, Loibl M, Gläsner J, Simon M, Rupp M, Grad S, et al. Vertebral osteomyelitis is characterised by increased RANK/OPG and RANKL/OPG expression ratios in vertebral bodies and intervertebral discs. Eur Cell Mater. 2021; 42:438-51. [DOI:10.22203/eCM.v042a27]
- Bax BE, Alam AS, Banerji B, Bax CM, Bevis PJ, Stevens CR, et al. Stimulation of osteoclastic bone resorption by hydrogen peroxide. Biochem Biophys Res Commun. 1992; 183(3):1153-8.[DOI:10.1016/S0006-291X(05)80311-0]
- Ha H, Kwak HB, Lee SW, Jin HM, Kim HM, Kim HH, et al. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp Cell Res. 2004; 301(2):119-27. [DOI:10.1016/j.yexcr.2004.07.035] [PMID]
- Rastogi R, Geng X, Li F, Ding Y. NOX Activation by subunit interaction and underlying mechanisms in disease. Front Cell Neurosci. 2017; 10:301. [DOI:10.3389/fncel.2016.00301]
- Nisimoto Y, Diebold BA, Cosentino-Gomes D, Lambeth JD. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014; 53(31):5111-20. [DOI:10.1021/bi500331y] [PMID]
- Van Asten SA, Nichols A, La Fontaine J, Bhavan K, Peters EJ, Lavery LA. The value of inflammatory markers to diagnose and monitor diabetic foot osteomyelitis. Int Wound J. 2017; 14(1):40-45. [DOI:10.1111/iwj.12545] [PMID]
- Mo M, Guilak F, Elward A, Quayle K, Thompson D, Brouillet K, et al. The use of biomarkers in the early diagnosis of septic arthritis and osteomyelitis-A pilot study. J Pediatr Orthop. 2022; 42(5):e526-32. [DOI:10.1097/BPO.0000000000002052]
- Bost KL, Bento JL, Petty CC, Schrum LW, Hudson MC, Marriott I. Monocyte chemoattractant protein-1 expression by osteoblasts following infection with Staphylococcus aureus or Salmonella. J Interferon Cytokine Res. 2001; 21(5):297-304. [DOI:10.1089/107999001300177484] [PMID]
- Marriott I, Gray DL, Rati DM, Fowler VG Jr, Stryjewski ME, Levin LS, et al. Osteoblasts produce monocyte chemoattractant protein-1 in a murine model of Staphylococcus aureus osteomyelitis and infected human bone tissue. Bone. 2005; 37(4):504-12. [DOI:10.1016/j.bone.2005.05.011] [PMID]
- van Spil WE, Drossaers-Bakker KW, Lafeber FP. Associations of CTX-II with biochemical markers of bone turnover raise questions on its tissue origin: Data from CHECK, a cohort study of early osteoarthritis. Ann Rheum Dis. 2013; 72(1):29-36. [DOI:10.1136/annrheumdis-2011-201177][PMID]
- Tekin Koruk S, Aksoy N, Hamidanoglu M, Karsen H, Unlu S, Bilinc H. The activity of paraoxonase and arylesterase in patients with osteomyelitis. Scand J Clin Lab Invest. 2012; 72(7):513-7. [DOI:10.3109/00365513.2012.700058]
- Duygu F, Tekin Koruk S, Aksoy N. Serum paraoxonase and arylesterase activities in various forms of hepatitis B virus infection. J Clin Lab Anal. 2011; 25(5):311-6.[DOI:10.1002/jcla.20473] [PMID]
- Soran N, Altindag O, Cakir H, Celik H, Demirkol A, Aksoy N. Assessment of paraoxonase activities in patients with knee osteoarthritis. Redox Rep. 2008; 13(5):194-8. [DOI:10.1179/135100008X308911] [PMID]
- Kralova J, Drobek A, Prochazka J, Spoutil F, Fabisik M, Glatzova D, et al. Dysregulated NADPH Oxidase Promotes Bone Damage in Murine Model of Autoinflammatory Osteomyelitis. J Immunol. 2020; 204(6):1607-20. [DOI:10.4049/jimmunol.1900953]
- Grbic R, Miric DJ, Kisic B, Popovic L, Nestorovic V, Vasic A. Sequential analysis of oxidative stress markers and vitamin C status in acute bacterial osteomyelitis. Mediators Inflamm. 2014; 2014:975061. [DOI:10.1155/2014/975061]
- Özkan S, Adanaş C, Demir C, Hakan H. The levels of oxidative DNA damage and some antioxidants in chronic osteomyelitis patients: A cross-sectional study. Int J Clin Pract. 2021; 75(11):e14704. [DOI:10.1111/ijcp.14704] [PMID]
- Jyoti A, Singh S, Mukhopadhyay B, Gavel R, Mishra SP. Free radicals and antioxidant status in chronic osteomyelitis patients: A case control study. J Clin Diagn Res. 2015; 9(4):BC08-10. [DOI:10.7860/JCDR/2015/11950.5781] [PMID]
- Gur M, Aslan M, Yildiz A, Demirbag R, Yilmaz R, Selek S, et al. Paraoxonase and arylesterase activities in coronary artery disease. Eur J Clin Invest. 2006; 36(11):779-87.[DOI:10.1111/j.1365-2362.2006.01727.x] [PMID]
- Cousins RJ. Absorption, transport, and hepatic metabolism of copper and zinc: Special reference to metallothionein and ceruloplasmin. Physiol Rev. 1985; 65(2):238-309. [DOI:10.1152/physrev.1985.65.2.238] [PMID]
- Fukai T, Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011; 15(6):1583-606. [DOI:10.1089/ars.2011.3999]
- Wu D, Liu B, Yin J, Xu T, Zhao S, Xu Q, et al. Detection of 8-hydroxydeoxyguanosine (8-OHdG) as a biomarker of oxidative damage in peripheral leukocyte DNA by UHPLC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2017; 1064:1-6. [DOI:10.1016/j.chroma.2017.03.005] [PMID]
- Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2' -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009; 27(2):120-39. [DOI:10.1080/10590500902885684]
Type of Study: Review Paper |
Subject:
Knee surgery
Received: 2024/02/2 | Accepted: 2024/03/18 | Published: 2024/05/1
Received: 2024/02/2 | Accepted: 2024/03/18 | Published: 2024/05/1
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |

Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
