

Volume 11, Issue 2 (May 2024)
JROS 2024, 11(2): 83-96 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Tabrizian P, Amroodi M N, Mokhtari K, Bahaeddini M, Amiri S. A Mini-review on Aging and Its Impact on Bone Health. JROS 2024; 11 (2) :83-96
URL: http://jros.iums.ac.ir/article-1-2278-en.html
URL: http://jros.iums.ac.ir/article-1-2278-en.html
Pouria Tabrizian1 

 , Morteza Nakhaei Amroodi1 , Khatere Mokhtari2 , Mohammadreza Bahaeddini1 , Saeedreza Amiri1
, Morteza Nakhaei Amroodi1 , Khatere Mokhtari2 , Mohammadreza Bahaeddini1 , Saeedreza Amiri1
, Morteza Nakhaei Amroodi1 , Khatere Mokhtari2 , Mohammadreza Bahaeddini1 , Saeedreza Amiri1
1- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Shafayahyaeian Hospital, Iran University of Medical Sciences, Tehran, Iran.
2- Department of Cell and Molecular Biology and Microbiology, Faculty of Biological Science and Technology, University of Isfahan, Isfahan, Iran.
2- Department of Cell and Molecular Biology and Microbiology, Faculty of Biological Science and Technology, University of Isfahan, Isfahan, Iran.
Full-Text [PDF 891 kb]
(269 Downloads)
| Abstract (HTML) (803 Views)
Full-Text: (407 Views)
Introduction
Aging and bone health
The aging process leads to the deterioration of bone composition, structure, and function [1, 2]. Given the strong association between age-related bone changes, research into the mechanisms underlying these alterations has expanded significantly in recent years.
Bone is a dynamic organ that performs both mechanical and homeostatic functions, constantly renewing itself through a process known as remodeling. The process of bone remodeling involves the degradation of old bone tissue, which is subsequently replaced by newly formed bone. This cyclical regeneration is critical for maintaining skeletal integrity and adapting to physiological demands. The activity occurs within localized regions of the bone, referred to as bone metabolic units (BMUs). These BMUs coordinate the actions of osteoclasts, which resorb bone, and osteoblasts, which synthesize new bone matrix, ensuring a balance between bone breakdown and formation. This tightly regulated process is essential for repairing microdamage, maintaining mineral homeostasis, and adapting to mechanical stress [3]. Within each BMU, the processes of bone formation by osteoblasts and bone resorption by osteoclasts are finely coordinated to maintain bone mass and structural integrity, enabling the bone to resist deformation. However, with aging, this balance is disrupted, resulting in an increase in bone resorption and a decrease in bone formation. This imbalance leads to a reduction in bone mass and strength, ultimately contributing to osteoporosis and a heightened risk of fractures.
Bone remodeling in aging bone
During the initial three decades of life, bone turnover is meticulously regulated to ensure equilibrium between bone resorption and formation, promoting optimal skeletal health. While turnover rates can vary among individuals, peak bone mass and bone size are typically achieved during adolescence and early adulthood. In women, this peak generally occurs between the ages of 15 and 20 years, marking a critical period for bone development and strength. Achieving optimal peak bone mass during this time is crucial, as it serves as a determinant of long-term bone health and resistance to age-related bone loss [4, 5].
In later life, menopause in women triggers a significant increase in bone resorption relative to formation, driven by decreased estrogen levels, which accelerates bone loss. Age-related bone loss is driven by the interplay of two opposing processes: Subperiosteal apposition, which occurs on the outer surface of the bone, and endosteal resorption, which takes place on the inner surface. With advancing age, the efficiency of bone remodeling diminishes, leading to a negative bone balance at specific BMU sites. After the fourth decade of life, periosteal bone formation begins to decline, while the activity of remodeling units on the endosteal surface increases. This shift leads to a progressive rise in endosteal resorption, which contributes to a reduction in bone mass and structural deterioration. These changes occur in both sexes, underscoring the importance of interventions to mitigate age-related bone loss and maintain skeletal health [6, 7]. Significant reductions in lumbar spine (LS) volumetric bone mineral density (vBMD), primarily attributed to trabecular bone loss in the vertebrae starting in the third decade, as well as a gradual decrease in cortical vBMD at the wrist, have been observed in both sexes as they age [8].
Secondary hyperparathyroidism
Elevated levels of serum parathyroid hormone (PTH) play a crucial role in regulating bone metabolism by stimulating osteoclastic activity, thereby enhancing bone resorption. This process predominantly affects cortical bone, leading to its gradual reduction in density and structural integrity. Over time, this progressive cortical bone loss weakens the overall mechanical strength of the skeleton, significantly increasing the risk of fractures. Furthermore, the sustained elevation of PTH, often associated with conditions such as primary hyperparathyroidism or age-related changes in calcium homeostasis, exacerbates these effects.
This condition underscores the importance of monitoring and managing PTH levels to preserve bone health and reduce fracture risk in vulnerable populations [9]. With advancing age, several factors can increase serum PTH levels, significantly impacting bone health. Key contributors include declining renal function, the use of medications such as loop diuretics (e.g. furosemide), and estrogen deficiency. In women, PTH secretion is initially moderated during the rapid phase of bone loss that occurs early in the postmenopausal period. However, as time progresses, PTH levels gradually rise, contributing to enhanced bone turnover. This age-related increase in PTH secretion underscores its role in the complex interplay of hormonal changes that drive alterations in bone remodeling and structural integrity [10].
PTH secretion also increases in aging men, following a pattern comparable to that observed in aging women. As men get older, a gradual rise in PTH levels occurs, contributing to changes in bone metabolism. This elevation in PTH is associated with reduced BMD and an increased risk of bone loss, reflecting similar age-related alterations in PTH regulation seen in women. The increase in PTH secretion in both sexes highlights its significant role in age-related bone remodeling processes [11, 12].
In aging men, normal circulating levels of gonadal sex steroids, such as testosterone, may offer some protective effect against bone resorption induced by elevated PTH levels. These sex steroids help mitigate the bone loss typically associated with increased PTH activity. As a result, it has been more challenging to establish a direct and unequivocal role for PTH in the development of age-related bone loss in men, as the influence of sex hormones appears to modulate the impact of elevated PTH on bone metabolism. This protective effect of gonadal hormones complicates the understanding of the precise mechanisms driving bone loss in older men [13].
Gonadal sex
It is well established that sex steroids, particularly estrogen and testosterone, are crucial for maintaining skeletal health. In women, the cessation of ovarian function during menopause leads to a significant decline in estrogen levels, which marks the onset of rapid bone loss. This reduction in estrogen triggers an imbalance in bone remodeling, favoring bone resorption over formation, and significantly increases the risk of osteoporosis and fractures. The decline in estrogen is a key factor in the accelerated bone loss observed in postmenopausal women, highlighting the importance of hormonal regulation in maintaining bone density and skeletal integrity throughout aging [14, 15].
The mechanisms underlying bone loss due to estrogen deficiency are complex, and the relative importance of each factor in the pathogenesis of this process remains unclear [16]. Generally, the impact of estrogen deficiency on bone occurs due to the loss of estrogen’s regulatory influence over bone resorption mediators. Estrogen typically suppresses osteoclast formation and activity by enhancing the production of osteoprotegerin (OPG) and transforming growth factor beta (TGF-β) [17, 18]. OPG acts as a soluble decoy receptor for the receptor activator of nuclear factor κB ligand (RANKL), thereby preventing osteoclast activation. Furthermore, TGF-β promotes osteoclast apoptosis, further aiding in the regulation of bone resorption [19]. Estrogen inhibits the production of RANKL by osteoblastic cells, as well as by T and B lymphocytes, thereby preventing osteoclast activation and bone resorption [18, 20]. Estrogen promotes apoptosis in osteoclast precursor cells and diminishes their differentiation by downregulating c-Jun activity, thereby inhibiting RANKL/macrophage colony-stimulating factor (M-CSF)-induced activating protein-1 (AP-1)-dependent transcription. This process contributes to the suppression of osteoclast formation and bone resorption [21, 22]. Estrogen may indirectly protect bone health by inhibiting the production of various bone-resorbing cytokines, including interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, M-CSF, and prostaglandins. These cytokines are key mediators of inflammation, which play a central role in promoting bone resorption. By reducing the levels of these inflammatory molecules, estrogen helps to mitigate the inflammatory processes that drive excessive bone breakdown, thereby contributing to the preservation of bone density and overall skeletal health [23-25].
It was previously believed that a decrease in serum testosterone in men was the primary driver of age-related bone loss. However, recent research has demonstrated that estrogen also plays a crucial role in this process. Similar to its effect in women, estrogen helps regulate bone metabolism in men, and its decline with aging contributes significantly to bone loss. The conversion of testosterone to estrogen in men is a crucial factor in maintaining bone density, underscoring the dual role of both sex steroids in skeletal health as men age. This knowledge has reshaped the understanding of age-related bone loss in men, emphasizing the importance of estrogen alongside testosterone [26-32]. Additionally, it has been found that testosterone, when coupled with high levels of sex hormone-binding globulin, is associated with a significantly increased fracture risk, even after adjusting for estradiol levels [33, 34].
Bone marrow fat
The accumulation of bone marrow fat is the main characteristic of age-related bone loss. This process occurs at the expense of osteoblastogenesis. As individuals age, the balance between adipogenesis and osteogenesis shifts, resulting in an increase in adipocyte production within the bone marrow while osteoblast activity declines. This shift not only contributes to reduced bone formation but also compromises bone density and strength, further exacerbating the risk of fractures and osteoporosis in older adults. The accumulation of bone marrow fat is thus considered a key marker of aging-related changes in the bone microenvironment [35]. The buildup of marrow fat seems to be an active process that occurs independently of estrogen, as it starts to develop in the third and fourth decades of life, long before the decline in estrogen levels seen with menopause. This finding indicates that factors beyond estrogen—such as age-related hormonal shifts and alterations in the bone microenvironment—play a role in the increased fat accumulation within the bone marrow. The progressive accumulation of marrow fat may disrupt normal bone remodeling, impairing osteoblast function and contributing further to age-related bone loss, even when there is no substantial drop in estrogen level [36-41]. Additionally, an inverse relationship exists between marrow fat volume and bone volume, a connection that is independent of sex and strongly correlates with the bone changes observed in individuals with osteoporosis. As marrow fat increases, there is a corresponding decrease in bone mass and density, reflecting the disruption of normal bone remodeling. This relationship highlights the potential role of marrow fat accumulation as both a marker and a contributor to the pathophysiology of osteoporosis, emphasizing its significance in the overall loss of bone strength and increased fracture risk associated with aging and osteoporotic conditions. This condition indicates that as marrow fat increases, bone volume decreases, contributing to the loss of bone density and strength typically associated with osteoporosis [42].
Mechanistically, mesenchymal stem cells (MSCs) appear to differentiate into adipocytes rather than osteoblasts preferentially. This shift in differentiation contributes to the accumulation of marrow fat and a diminished capacity for osteoblastogenesis, ultimately resulting in decreased bone formation and increased bone loss during aging [43]. Factors such as reduced growth factor availability, compromised blood supply, and changes in oxygen tension within the bone marrow drive this shift, leading to increased marrow fat accumulation and decreased bone formation, which are key characteristics of age-related bone loss [44]. Moreover, the primary lineage-specific transcription factors that regulate MSC differentiation are Runx2, which drives osteoblastogenesis, and peroxisome proliferator-activated receptor gamma 2 (PPARγ2), which governs adipogenesis. Runx2 is essential for the commitment of MSCs to the osteoblastic lineage, promoting bone formation. At the same time, PPARγ2 plays a pivotal role in directing MSCs toward adipocyte differentiation, leading to the accumulation of marrow fat. The balance between these two transcription factors is crucial for maintaining normal bone homeostasis, and disruptions in this balance can contribute to pathological conditions such as osteoporosis, where excessive adipogenesis may impair bone formation. Runx2 is crucial for osteoblast differentiation and bone formation, while PPARγ2 is a key regulator of adipocyte differentiation. The balance between these transcription factors is critical in determining whether MSCs differentiate into osteoblasts or adipocytes. Age-related changes can disrupt this balance, promoting adipogenesis and contributing to bone loss [35, 45]. As individuals age, there is a shift toward increased expression of PPARγ2 in MSCs, accompanied by a concurrent decrease in Runx2 expression. This alteration results in diminished osteoblast differentiation and a heightened tendency for MSCs to differentiate into adipocytes rather than osteoblasts. As a consequence, bone formation is hindered, and marrow fat accumulation rises, contributing to the overall reduction in bone mass and quality associated with aging [46].
Aging itself, independent of hormonal changes, appears to play a crucial role in the process of bone marrow adipogenesis. Thus, senile osteoporosis may represent a form of lipotoxicity, where the accumulation of adipocytes in the bone marrow disrupts normal bone remodeling. In this scenario, the excessive bone marrow fat interferes with osteoblast function, leading to a decline in bone formation and contributing to the development of osteoporosis. This finding highlights the importance of investigating the underlying mechanisms by which aging-induced adipogenesis influences bone health and the pathogenesis of age-related bone loss [47]. Indeed, bone marrow adipocytes seem to have a toxic effect on osteoblasts. The accumulation of fat within the bone marrow creates an environment that impairs osteoblast function, potentially through the release of fatty acids or inflammatory cytokines. This disruption in osteoblast activity hinders bone formation, contributing to the imbalance between bone resorption and formation seen in conditions like osteoporosis. The toxic influence of marrow adipocytes on osteoblasts highlights the complex interplay between adipogenesis and osteogenesis in the pathophysiology of age-related bone loss [48]. Cocultures of adipocytes and osteoblasts have demonstrated that adipocytes inhibit osteoblast activity and survival, likely through the secretion of adipokines and fatty acids. The increased number of adipocytes within the bone marrow creates a detrimental microenvironment, where these molecules disrupt osteoblast function. Adipokines, which are signaling proteins produced by adipocytes, and fatty acids released by the growing adipocyte population can directly interfere with osteoblast differentiation, activity, and overall bone formation. This interaction disrupts the balance between bone resorption and formation, leading to accelerated bone loss in conditions like osteoporosis [49].
Other factors
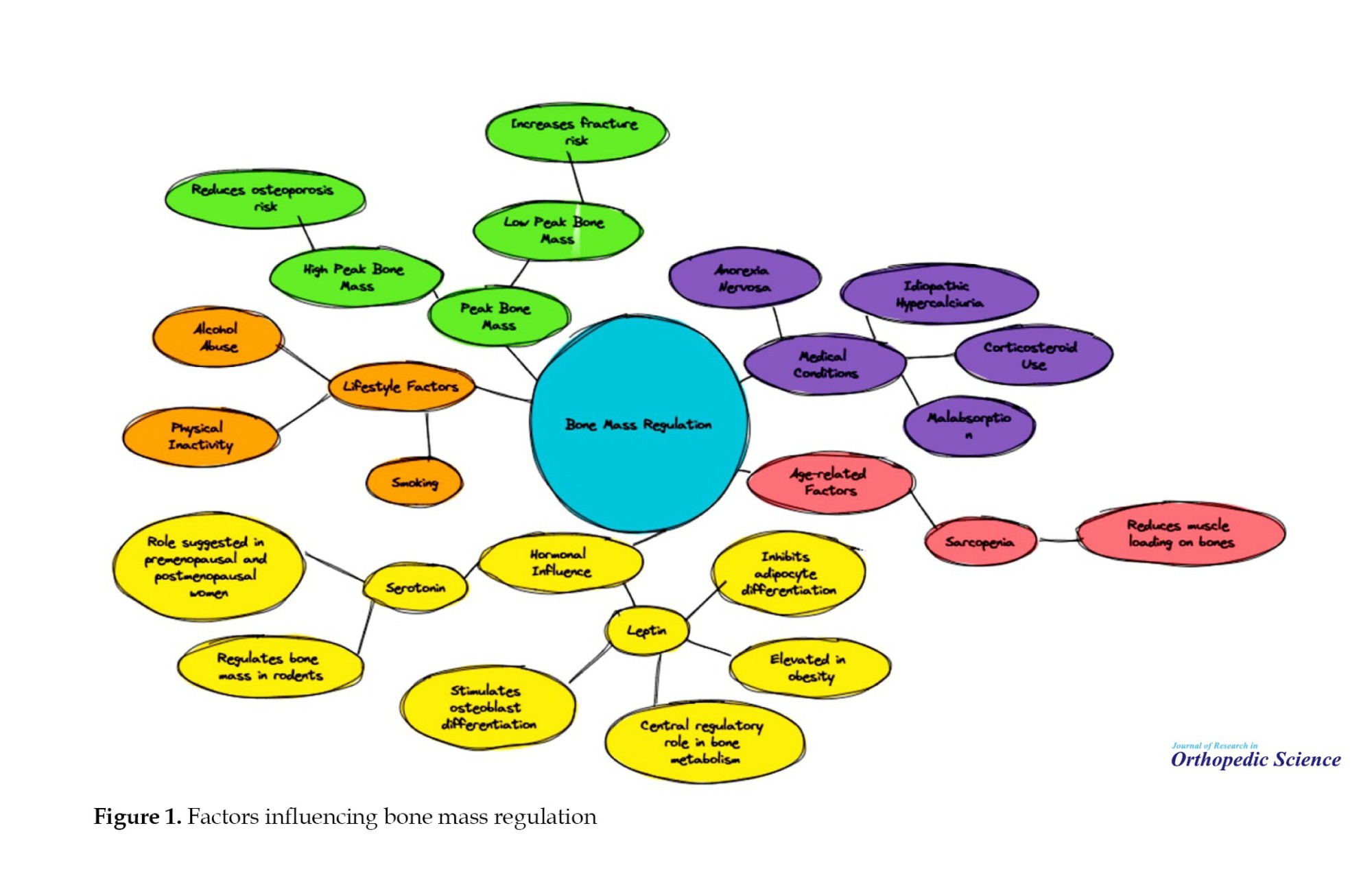
Several clinical studies have highlighted the intricate relationships between body fat, bone mass, and regulatory hormones such as leptin and serotonin. Leptin, a hormone elevated in obesity, is closely associated with fat mass and has been found to influence bone metabolism through its effects on osteoblast differentiation and neuronal pathways. Similarly, serotonin has shown potential regulatory effects on bone mass in both animal and human studies. The development of peak bone mass is a critical determinant of age-related bone loss, with higher peak bone mass serving as a protective factor against osteoporosis. Other contributors to fracture risk include medical conditions, behavioral factors, and possibly sarcopenia, which can reduce muscle loading on the bone [50-60]. Additional details are provided in Figure 1.
Exercise
Aging is associated with decreased physical activity and mechanical loading, resulting in reduced mechanical stimulation of osteoblasts. As a consequence, osteoblasts show a diminished secretion of OPG and an increased production of receptor activator of RANKL, as well as pro-inflammatory cytokines such as IL-1, IL-6, IL-11, and TNF-α. These alterations promote the formation and activation of osteoclasts, leading to increased bone resorption. The imbalance between osteoblast and osteoclast activity contributes to the acceleration of age-related bone loss, heightening the risk of osteoporosis and fractures. Furthermore, the decline in OPG levels facilitates increased interaction between RANKL and the receptor activator of nuclear factor κB (RANK), thereby amplifying osteoclastogenesis and bone resorption [47, 61]. Recent studies have demonstrated that the anabolic response to exercise is linked to the presence of Lamin A/C [62]. In humans, functional loading has been shown to enhance bone mass, with exercise training programs able to prevent or reverse about 1% of annual bone loss in the LS and femoral neck (FN) in both premenopausal and postmenopausal women. This finding highlights the significant role that mechanical loading, such as weight-bearing exercises, plays in maintaining bone health. Regular physical activity helps stimulate bone remodeling, promoting the maintenance of bone density and reducing the risk of osteoporosis and fractures. This effect highlights the importance of exercise in both preventing and mitigating age-related bone loss in women across various life stages [63, 64].
Bone mechanical behavior
The components of bone are precisely balanced to provide resistance to fractures while minimizing skeletal weight. Stiffness, which refers to the bone’s resistance to deformation, and strength, the maximum stress a bone can withstand before failure, are essential for supporting substantial loads and maintaining structural integrity. At the same time, toughness, or ductility, plays a critical role in absorbing impact energy, allowing bones to withstand forces without fracturing. This intricate balance between stiffness, strength, and toughness is crucial in ensuring that bones can both support mechanical demands and absorb stresses encountered during daily activities, thereby reducing the likelihood of injury. A shift in this balance toward higher tissue mineral content typically results in stiffer but more brittle bones.
Furthermore, changes in collagen structure can increase brittleness, as modifications in cross-linking not only stiffen the organic matrix but also alter the morphology of the mineral component, a topic explored further in this discussion. Notably, while BMD decreases in conditions such as osteoporosis, it increases in disorders like osteopetrosis [65-67]. BMD is widely regarded by researchers and clinicians as a key marker of fracture susceptibility, particularly due to its age-related decline in both men and women. Understanding the mechanisms behind the increased fragility and brittleness of bone with age is therefore essential. Numerous studies have demonstrated that cortical bone becomes progressively more brittle and weaker as individuals grow older [68-70].
Bone morphology
Morphology refers to the shape and structure of bones, which can be classified into several categories based on their form and function. These categories include long bones, such as the femur and tibia, which are primarily responsible for support and movement; short bones, like those found in the hands and feet, which provide stability and support with limited motion; and flat bones, such as the calvaria (skull) or sternum, which serve protective roles and provide a broad surface for muscle attachment. Each bone type is adapted to its specific functional requirements, contributing to the overall efficiency and resilience of the skeletal system. The morphological characteristics that influence bone strength include the size and shape of the bones [71-74].
During both development and aging, bone shape adapts in response to mechanical load, a process described by Wolff’s Law. According to this principle, bones remodel and adjust their structure in response to the stresses they experience, with increased load leading to bone formation and reduced load resulting in bone resorption. As bones undergo changes in shape—such as lengthening, thinning of their walls, or altering their center of gravity—their functional capabilities are also modified. These adaptations allow the skeletal system to optimize its strength and efficiency in response to changing mechanical demands, ensuring that bones remain resilient and capable of supporting the body’s activities throughout life [75-79]. A recent study examining the trabecular bones in the proximal tibias of 23-month-old and 5-month-old rats found a significant decrease in mineral density, bone volume fraction, and trabecular number in the older rats compared to the younger ones. These results underscore the impact of aging on bone structure, indicating that as rats age, there is a notable deterioration in critical bone characteristics that contribute to overall bone strength. The reduction in mineral density and trabecular number observed in the older rats reflects a decline in bone mass and structural integrity, which may increase the risk of fractures and other age-related bone pathologies [80].
In necropsied tibias from males aged 17 to 46 years, significant alterations in mechanical properties were found to correlate with the tibia’s size (slenderness), independent of age. As individuals aged, there was a noticeable increase in mechanical weakening, accompanied by more brittle bone behavior. These findings suggest that the structural changes in the tibia, particularly related to its size, contribute to the decline in mechanical strength over time, leading to increased fragility and a higher risk of fractures as age progresses [81].
During development, long bones undergo significant morphological changes. In the early stages of life, the cross-sectional geometry of these bones is characterized by a relatively uniform outer wall thickness. As the individual matures, this structure gradually transforms into a more ellipsoidal shape through a process known as “cortical drift.” This process involves remodeling the bone’s cortex, where the outer layer thickens and shifts, optimizing its shape and strength in response to mechanical forces. The resulting bone structure is more efficient in supporting the body’s weight and movement, adapting to the functional demands placed on the skeleton over time [82]. As with other transformations, bone geometry continuously changes throughout life in response to mechanical forces and biological signaling [82].
Bone cells
Bone is composed of several distinct cell types [83-86]. Osteoblastogenesis is regulated by various signaling pathways [87, 88]. Some osteoblasts differentiate into small, inactive bone-lining cells along dormant surfaces, while the remaining osteoblasts become embedded in mineralized matrix and develop long dendritic processes [88-90]. Connexin 43, the most prevalent gap junction protein in bone, plays a crucial role in bone modeling and remodeling [89, 90].
Osteoclast differentiation is reduced in older animals compared to younger ones, resulting in bone resorption rates that significantly exceed the rates of new bone formation. This imbalance between resorption and formation contributes to the progressive loss of bone mass and density, which is a hallmark of age-related bone degeneration. The impaired differentiation of osteoclasts in older animals may lead to an inefficient response to bone remodeling stimuli, further exacerbating the disparity between bone resorption and the ability to form new bone tissue [80, 91-93].
Osteoblasts, osteoclasts, and osteocytes have a limited lifespan, which is controlled by both intrinsic replication cycles and external influences. The Hayflick limit refers to the principle that cells can divide only a set number of times, with this replicative capacity gradually declining with age. This limitation is thought to be a result of telomere shortening during each cell division, leading to cellular senescence or apoptosis once the telomeres reach a critically short length. In bone cells, this reduction in proliferative capacity over time may contribute to decreased bone regeneration and impaired bone remodeling in older individuals [94]. The activity of telomerase is not infinite and decreases with age, resulting in a gradual reduction in the regenerative potential of stem cells over time [95]. Telomere damage induced by UV light and oxidative stress can hasten telomere shortening, thereby reducing cell lifespan. This heightened damage accelerates cellular aging and may play a significant role in the progressive decline of tissue regeneration and function over time [96].
Werner syndrome in humans is a genetic disorder that leads to the premature onset of age-related diseases, characterized by accelerated telomere shortening after puberty. This syndrome results from mutations in the WRN gene, which encodes a helicase involved in maintaining genomic stability. As a consequence, individuals with Werner syndrome experience accelerated cellular aging, with telomeres shortening at a significantly faster rate than in the general population. This rapid telomere attrition contributes to the early onset of conditions typically associated with aging, such as osteoporosis, cardiovascular diseases, and diabetes, underscoring the critical role of telomere maintenance in aging and age-related pathologies. This rapid telomere attrition contributes to the early development of aging-related conditions, including atherosclerosis, osteoporosis, and cancer [97, 98].
Cells often undergo changes in gene expression and activity with age, which may be attributed to alterations in gene (mRNA) translation. A study has demonstrated that treatments aimed at reducing mRNA translation in animals can extend their lifespan, implying that modulating protein synthesis could have an impact on aging and longevity [99]. Aging is particularly significant for bone cells, though it affects all cell types, as it is associated with a reduced ability to respond effectively to mechanical forces. This diminished responsiveness can negatively impact bone health and the regulation of bone remodeling, contributing to age-related skeletal problems [78].
Among the key cellular changes that occur with aging, contributing to the decline in bone’s mechanical function, are alterations in both the amount and rate of bone remodeling. Bone remodeling is a dynamic process that involves the resorption of old bone by osteoclasts, followed by the formation of new bone by osteoblasts. As individuals age, this process becomes less efficient, with a decrease in osteoblast activity and an increase in osteoclast activity. This imbalance results in a net loss of bone mass and changes in bone architecture, leading to decreased bone strength and increased fragility. Additionally, the reduced ability of osteoblasts to effectively regenerate bone tissue contributes to the overall decline in skeletal integrity, making bones more susceptible to fractures and other age-related conditions. With age, the amount of bone deposited during each remodeling cycle diminishes. This reduction may be attributed to a decline in the number of osteoblast precursors, a decrease in the stem cell population from which these precursors are derived, or a shortening of the lifespan of osteoblasts [100]. The signals that regulate the differentiation of osteoblast precursors decline with age, which likely contributes to the reduction in osteoblast numbers. These signaling pathways, including factors such as bone morphogenetic proteins (BMPs) and the Wnt/β-catenin signaling pathway, are essential for promoting the maturation of osteoblasts from their precursor cells. As these pathways become less effective with aging, the production of osteoblasts diminishes, leading to a decrease in bone formation. This reduction in osteoblast activity can contribute to the overall decline in bone mass and the increased risk of fractures typically observed in older individuals. This diminished signaling further impairs bone formation, leading to a decline in bone density and mechanical strength associated with aging [101]. In non-human primates, the number of hematopoietic cells, which serve as osteoclast precursors, decreases with age [100, 101].
Cell senescence may alter how cells respond to apoptotic signals, potentially influencing the way bone cells react to stress or damage. However, whether this occurs specifically in bone cells is an area that requires further investigation. Understanding the influence of senescence on bone cell function could offer valuable insights into age-related bone diseases and the mechanisms driving bone fragility [96].
Signaling pathways in bone cell aging and their impact on bone health
Various signaling pathways play a crucial role in regulating the differentiation and metabolism of bone cells, maintaining the delicate balance between bone formation and resorption. Among these, the Wnt/β-catenin pathway is essential for osteoblast differentiation and bone formation, while the RANK/RANKL/OPG signaling axis governs osteoclast differentiation and bone resorption. Additionally, BMPs are key regulators of osteoblast differentiation, and pathways such as Notch and Hedgehog further modulate bone cell activity. These interconnected pathways not only oversee the development of osteoblasts, osteoclasts, and osteocytes but also influence their function and survival, ensuring skeletal homeostasis. Disruptions in these signaling mechanisms can lead to bone-related disorders, such as osteoporosis, highlighting their importance in maintaining bone health [93, 102].
Signaling through a family of genes known as Wnt is thought to regulate cellular aging across various cell types. However, the exact mechanism by which Wnt signaling influences aging remains a topic of ongoing research and debate. The role of Wnt pathways in aging may involve regulating cellular processes such as proliferation, differentiation, and apoptosis, all of which are essential for maintaining bone homeostasis [103-106]. The findings suggest that sustaining or augmenting Wnt signaling may offer a potential approach to mitigate age-related bone loss and promote bone health in older individuals [107]. It suggests that increased levels of sFRP-4 may contribute to reduced bone mass and a higher risk of osteoporosis with aging, possibly by interfering with the regulation of osteoblast activity and bone formation [108].
Recent studies have suggested that oxidative stress may play a significant role in the aging of bone cells, with effects mediated indirectly through the Wnt signaling pathway. Reactive oxygen species are thought to activate FoxO transcription factors, which then bind to β-catenin. This interaction may disrupt the normal function of β-catenin, impairing osteoblast differentiation and function. As a result, oxidative stress may contribute to an imbalance in bone remodeling, potentially accelerating bone loss and increasing the risk of age-related bone diseases such as osteoporosis. This binding reduces the effective concentration of β-catenin in cells, subsequently impairing osteoblast formation. By disrupting the Wnt signaling pathway, oxidative stress hinders osteoblast differentiation and bone formation, thereby contributing to age-related bone loss [109, 110]. The reduction in Wnt signaling, triggered by PPAR-γ activation through ligands produced during lipid oxidation, also plays a role in the age-related decrease in osteoblast formation. This reduction in Wnt signaling impairs the differentiation and function of osteoblasts, thereby disrupting the balance between bone formation and resorption. As a result, the ability to regenerate bone tissue is diminished, leading to a decrease in bone mass and increased susceptibility to fractures with aging. This process exacerbates the bone cells’ inability to maintain bone mass and function with age. The activation of PPAR-γ, typically associated with adipogenesis, inhibits osteoblast differentiation, thereby disrupting the balance between bone resorption and formation. This imbalance results in weakened bone structure and an increased risk of fractures [65].
Bone protein
The organic matrix of bone is primarily composed of collagen, with type I collagen accounting for the majority, along with approximately 5% non-collagenous proteins [111]. Collagen cross-links exhibit distinct changes with aging, affecting the structural integrity and mechanical properties of the bone [112-114]. Building on this finding, the authors speculated that as aging progresses, the rate of nonenzymatic cross-linking increases, while the formation of mature enzymatic cross-links decreases. These alterations could reduce bone strength and toughness, ultimately compromising the bone’s ability to resist crack propagation. Similar findings have also been reported in bovine bone [115]. The accumulation of advanced glycation end products promotes osteoclast activity while inhibiting osteoblast differentiation, thereby contributing to the increased bone fragility associated with aging [116]. As tissue ages, the orientation of collagen fibers becomes more aligned, which can affect the mechanical properties of the bone [117]. With older age, changes occur in the expression and relative abundance of non-collagenous proteins in bone, which can impact its structural integrity and function [118, 119]. It is important to note that with age, not only do the distributions of non-collagenous proteins in bone change, but the extent of their post-translational modifications also diminishes [120, 121]. A decline in protein production with age has been documented, alongside an increase in bone matrix protein fragments in the older group [122].
Minerals
Bone mineral content, commonly known as “mineralization” or “ash content,” tends to increase with age. Classical research has shown that bone-breaking stress increases exponentially with higher ash content. However, once the ash content reaches its maximum level, bone toughness—the ability to resist fracture—begins to decline. This condition indicates that while greater mineralization strengthens the bone against breaking forces, excessive mineralization can lead to increased brittleness, thereby reducing the bone’s ability to absorb impact and making it more susceptible to fractures. This interplay between strength and toughness is essential for maintaining bone integrity over time [123].
The mineral present in bone is a form of hydroxyapatite, a naturally occurring mineral with the chemical formula [Ca10(PO4)6(OH)2]. Bone mineral crystals are nanoscale structures, typically measuring about 1 to 1.5 nm in thickness, 5 to 25 nm in width, and 8 to 40 nm in length. These nanoscale dimensions enable the unique mechanical properties of bone, including its strength and flexibility, which contribute to its ability to support and protect the body while maintaining a light and resilient structure [124-126]. These bone mineral crystals also incorporate various inclusions and substitutions that evolve with age. A notable substitution is carbonate, which replaces hydroxyl and phosphate groups within the surface and crystal lattice of apatite. This alteration can impact the structural characteristics of the crystals, potentially modifying their mechanical properties. As aging progresses, these changes in the mineral composition may affect bone mineralization, contributing to the decline in bone strength and an increased risk of fractures [127-129].
Conclusion
The aging process significantly disrupts bone remodeling, leading to a decline in bone mass, strength, and structural integrity. Key factors, including hormonal imbalances, elevated PTH levels, and the accumulation of bone marrow fat, contribute to the deterioration of bone health. Additionally, changes in bone cell functionality and alterations in the bone matrix and mineral composition further compromise bone strength and toughness. As these changes accelerate the risk of osteoporosis and fractures, understanding the molecular and cellular mechanisms underlying bone aging is crucial for developing effective therapeutic approaches. Continued research is essential for identifying novel interventions that can mitigate the adverse effects of aging on bone health and enhance the quality of life in the aging population.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This study was supported by the Iran University of Medical Sciences, Tehran, Iran.
Authors' contributions
Conceptualization and supervision: Saeedreza Amiri; Methodology: Pouria Tabrizian, and Morteza Nakhaei Amroodi; Data collection: Khatere Mokhtari, and Mohammadreza Bahaeddini; Data analysis: Khatere Mokhtari and Mohammadreza Bahaeddini; Investigation and writing: All authors; Funding acquisition and Resources: Saeedreza Amiri.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors wish to thank all individuals who helped us develop this research.
References
Aging and bone health
The aging process leads to the deterioration of bone composition, structure, and function [1, 2]. Given the strong association between age-related bone changes, research into the mechanisms underlying these alterations has expanded significantly in recent years.
Bone is a dynamic organ that performs both mechanical and homeostatic functions, constantly renewing itself through a process known as remodeling. The process of bone remodeling involves the degradation of old bone tissue, which is subsequently replaced by newly formed bone. This cyclical regeneration is critical for maintaining skeletal integrity and adapting to physiological demands. The activity occurs within localized regions of the bone, referred to as bone metabolic units (BMUs). These BMUs coordinate the actions of osteoclasts, which resorb bone, and osteoblasts, which synthesize new bone matrix, ensuring a balance between bone breakdown and formation. This tightly regulated process is essential for repairing microdamage, maintaining mineral homeostasis, and adapting to mechanical stress [3]. Within each BMU, the processes of bone formation by osteoblasts and bone resorption by osteoclasts are finely coordinated to maintain bone mass and structural integrity, enabling the bone to resist deformation. However, with aging, this balance is disrupted, resulting in an increase in bone resorption and a decrease in bone formation. This imbalance leads to a reduction in bone mass and strength, ultimately contributing to osteoporosis and a heightened risk of fractures.
Bone remodeling in aging bone
During the initial three decades of life, bone turnover is meticulously regulated to ensure equilibrium between bone resorption and formation, promoting optimal skeletal health. While turnover rates can vary among individuals, peak bone mass and bone size are typically achieved during adolescence and early adulthood. In women, this peak generally occurs between the ages of 15 and 20 years, marking a critical period for bone development and strength. Achieving optimal peak bone mass during this time is crucial, as it serves as a determinant of long-term bone health and resistance to age-related bone loss [4, 5].
In later life, menopause in women triggers a significant increase in bone resorption relative to formation, driven by decreased estrogen levels, which accelerates bone loss. Age-related bone loss is driven by the interplay of two opposing processes: Subperiosteal apposition, which occurs on the outer surface of the bone, and endosteal resorption, which takes place on the inner surface. With advancing age, the efficiency of bone remodeling diminishes, leading to a negative bone balance at specific BMU sites. After the fourth decade of life, periosteal bone formation begins to decline, while the activity of remodeling units on the endosteal surface increases. This shift leads to a progressive rise in endosteal resorption, which contributes to a reduction in bone mass and structural deterioration. These changes occur in both sexes, underscoring the importance of interventions to mitigate age-related bone loss and maintain skeletal health [6, 7]. Significant reductions in lumbar spine (LS) volumetric bone mineral density (vBMD), primarily attributed to trabecular bone loss in the vertebrae starting in the third decade, as well as a gradual decrease in cortical vBMD at the wrist, have been observed in both sexes as they age [8].
Secondary hyperparathyroidism
Elevated levels of serum parathyroid hormone (PTH) play a crucial role in regulating bone metabolism by stimulating osteoclastic activity, thereby enhancing bone resorption. This process predominantly affects cortical bone, leading to its gradual reduction in density and structural integrity. Over time, this progressive cortical bone loss weakens the overall mechanical strength of the skeleton, significantly increasing the risk of fractures. Furthermore, the sustained elevation of PTH, often associated with conditions such as primary hyperparathyroidism or age-related changes in calcium homeostasis, exacerbates these effects.
This condition underscores the importance of monitoring and managing PTH levels to preserve bone health and reduce fracture risk in vulnerable populations [9]. With advancing age, several factors can increase serum PTH levels, significantly impacting bone health. Key contributors include declining renal function, the use of medications such as loop diuretics (e.g. furosemide), and estrogen deficiency. In women, PTH secretion is initially moderated during the rapid phase of bone loss that occurs early in the postmenopausal period. However, as time progresses, PTH levels gradually rise, contributing to enhanced bone turnover. This age-related increase in PTH secretion underscores its role in the complex interplay of hormonal changes that drive alterations in bone remodeling and structural integrity [10].
PTH secretion also increases in aging men, following a pattern comparable to that observed in aging women. As men get older, a gradual rise in PTH levels occurs, contributing to changes in bone metabolism. This elevation in PTH is associated with reduced BMD and an increased risk of bone loss, reflecting similar age-related alterations in PTH regulation seen in women. The increase in PTH secretion in both sexes highlights its significant role in age-related bone remodeling processes [11, 12].
In aging men, normal circulating levels of gonadal sex steroids, such as testosterone, may offer some protective effect against bone resorption induced by elevated PTH levels. These sex steroids help mitigate the bone loss typically associated with increased PTH activity. As a result, it has been more challenging to establish a direct and unequivocal role for PTH in the development of age-related bone loss in men, as the influence of sex hormones appears to modulate the impact of elevated PTH on bone metabolism. This protective effect of gonadal hormones complicates the understanding of the precise mechanisms driving bone loss in older men [13].
Gonadal sex
It is well established that sex steroids, particularly estrogen and testosterone, are crucial for maintaining skeletal health. In women, the cessation of ovarian function during menopause leads to a significant decline in estrogen levels, which marks the onset of rapid bone loss. This reduction in estrogen triggers an imbalance in bone remodeling, favoring bone resorption over formation, and significantly increases the risk of osteoporosis and fractures. The decline in estrogen is a key factor in the accelerated bone loss observed in postmenopausal women, highlighting the importance of hormonal regulation in maintaining bone density and skeletal integrity throughout aging [14, 15].
The mechanisms underlying bone loss due to estrogen deficiency are complex, and the relative importance of each factor in the pathogenesis of this process remains unclear [16]. Generally, the impact of estrogen deficiency on bone occurs due to the loss of estrogen’s regulatory influence over bone resorption mediators. Estrogen typically suppresses osteoclast formation and activity by enhancing the production of osteoprotegerin (OPG) and transforming growth factor beta (TGF-β) [17, 18]. OPG acts as a soluble decoy receptor for the receptor activator of nuclear factor κB ligand (RANKL), thereby preventing osteoclast activation. Furthermore, TGF-β promotes osteoclast apoptosis, further aiding in the regulation of bone resorption [19]. Estrogen inhibits the production of RANKL by osteoblastic cells, as well as by T and B lymphocytes, thereby preventing osteoclast activation and bone resorption [18, 20]. Estrogen promotes apoptosis in osteoclast precursor cells and diminishes their differentiation by downregulating c-Jun activity, thereby inhibiting RANKL/macrophage colony-stimulating factor (M-CSF)-induced activating protein-1 (AP-1)-dependent transcription. This process contributes to the suppression of osteoclast formation and bone resorption [21, 22]. Estrogen may indirectly protect bone health by inhibiting the production of various bone-resorbing cytokines, including interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, M-CSF, and prostaglandins. These cytokines are key mediators of inflammation, which play a central role in promoting bone resorption. By reducing the levels of these inflammatory molecules, estrogen helps to mitigate the inflammatory processes that drive excessive bone breakdown, thereby contributing to the preservation of bone density and overall skeletal health [23-25].
It was previously believed that a decrease in serum testosterone in men was the primary driver of age-related bone loss. However, recent research has demonstrated that estrogen also plays a crucial role in this process. Similar to its effect in women, estrogen helps regulate bone metabolism in men, and its decline with aging contributes significantly to bone loss. The conversion of testosterone to estrogen in men is a crucial factor in maintaining bone density, underscoring the dual role of both sex steroids in skeletal health as men age. This knowledge has reshaped the understanding of age-related bone loss in men, emphasizing the importance of estrogen alongside testosterone [26-32]. Additionally, it has been found that testosterone, when coupled with high levels of sex hormone-binding globulin, is associated with a significantly increased fracture risk, even after adjusting for estradiol levels [33, 34].
Bone marrow fat
The accumulation of bone marrow fat is the main characteristic of age-related bone loss. This process occurs at the expense of osteoblastogenesis. As individuals age, the balance between adipogenesis and osteogenesis shifts, resulting in an increase in adipocyte production within the bone marrow while osteoblast activity declines. This shift not only contributes to reduced bone formation but also compromises bone density and strength, further exacerbating the risk of fractures and osteoporosis in older adults. The accumulation of bone marrow fat is thus considered a key marker of aging-related changes in the bone microenvironment [35]. The buildup of marrow fat seems to be an active process that occurs independently of estrogen, as it starts to develop in the third and fourth decades of life, long before the decline in estrogen levels seen with menopause. This finding indicates that factors beyond estrogen—such as age-related hormonal shifts and alterations in the bone microenvironment—play a role in the increased fat accumulation within the bone marrow. The progressive accumulation of marrow fat may disrupt normal bone remodeling, impairing osteoblast function and contributing further to age-related bone loss, even when there is no substantial drop in estrogen level [36-41]. Additionally, an inverse relationship exists between marrow fat volume and bone volume, a connection that is independent of sex and strongly correlates with the bone changes observed in individuals with osteoporosis. As marrow fat increases, there is a corresponding decrease in bone mass and density, reflecting the disruption of normal bone remodeling. This relationship highlights the potential role of marrow fat accumulation as both a marker and a contributor to the pathophysiology of osteoporosis, emphasizing its significance in the overall loss of bone strength and increased fracture risk associated with aging and osteoporotic conditions. This condition indicates that as marrow fat increases, bone volume decreases, contributing to the loss of bone density and strength typically associated with osteoporosis [42].
Mechanistically, mesenchymal stem cells (MSCs) appear to differentiate into adipocytes rather than osteoblasts preferentially. This shift in differentiation contributes to the accumulation of marrow fat and a diminished capacity for osteoblastogenesis, ultimately resulting in decreased bone formation and increased bone loss during aging [43]. Factors such as reduced growth factor availability, compromised blood supply, and changes in oxygen tension within the bone marrow drive this shift, leading to increased marrow fat accumulation and decreased bone formation, which are key characteristics of age-related bone loss [44]. Moreover, the primary lineage-specific transcription factors that regulate MSC differentiation are Runx2, which drives osteoblastogenesis, and peroxisome proliferator-activated receptor gamma 2 (PPARγ2), which governs adipogenesis. Runx2 is essential for the commitment of MSCs to the osteoblastic lineage, promoting bone formation. At the same time, PPARγ2 plays a pivotal role in directing MSCs toward adipocyte differentiation, leading to the accumulation of marrow fat. The balance between these two transcription factors is crucial for maintaining normal bone homeostasis, and disruptions in this balance can contribute to pathological conditions such as osteoporosis, where excessive adipogenesis may impair bone formation. Runx2 is crucial for osteoblast differentiation and bone formation, while PPARγ2 is a key regulator of adipocyte differentiation. The balance between these transcription factors is critical in determining whether MSCs differentiate into osteoblasts or adipocytes. Age-related changes can disrupt this balance, promoting adipogenesis and contributing to bone loss [35, 45]. As individuals age, there is a shift toward increased expression of PPARγ2 in MSCs, accompanied by a concurrent decrease in Runx2 expression. This alteration results in diminished osteoblast differentiation and a heightened tendency for MSCs to differentiate into adipocytes rather than osteoblasts. As a consequence, bone formation is hindered, and marrow fat accumulation rises, contributing to the overall reduction in bone mass and quality associated with aging [46].
Aging itself, independent of hormonal changes, appears to play a crucial role in the process of bone marrow adipogenesis. Thus, senile osteoporosis may represent a form of lipotoxicity, where the accumulation of adipocytes in the bone marrow disrupts normal bone remodeling. In this scenario, the excessive bone marrow fat interferes with osteoblast function, leading to a decline in bone formation and contributing to the development of osteoporosis. This finding highlights the importance of investigating the underlying mechanisms by which aging-induced adipogenesis influences bone health and the pathogenesis of age-related bone loss [47]. Indeed, bone marrow adipocytes seem to have a toxic effect on osteoblasts. The accumulation of fat within the bone marrow creates an environment that impairs osteoblast function, potentially through the release of fatty acids or inflammatory cytokines. This disruption in osteoblast activity hinders bone formation, contributing to the imbalance between bone resorption and formation seen in conditions like osteoporosis. The toxic influence of marrow adipocytes on osteoblasts highlights the complex interplay between adipogenesis and osteogenesis in the pathophysiology of age-related bone loss [48]. Cocultures of adipocytes and osteoblasts have demonstrated that adipocytes inhibit osteoblast activity and survival, likely through the secretion of adipokines and fatty acids. The increased number of adipocytes within the bone marrow creates a detrimental microenvironment, where these molecules disrupt osteoblast function. Adipokines, which are signaling proteins produced by adipocytes, and fatty acids released by the growing adipocyte population can directly interfere with osteoblast differentiation, activity, and overall bone formation. This interaction disrupts the balance between bone resorption and formation, leading to accelerated bone loss in conditions like osteoporosis [49].
Other factors
Several clinical studies have highlighted the intricate relationships between body fat, bone mass, and regulatory hormones such as leptin and serotonin. Leptin, a hormone elevated in obesity, is closely associated with fat mass and has been found to influence bone metabolism through its effects on osteoblast differentiation and neuronal pathways. Similarly, serotonin has shown potential regulatory effects on bone mass in both animal and human studies. The development of peak bone mass is a critical determinant of age-related bone loss, with higher peak bone mass serving as a protective factor against osteoporosis. Other contributors to fracture risk include medical conditions, behavioral factors, and possibly sarcopenia, which can reduce muscle loading on the bone [50-60]. Additional details are provided in Figure 1.
Exercise
Aging is associated with decreased physical activity and mechanical loading, resulting in reduced mechanical stimulation of osteoblasts. As a consequence, osteoblasts show a diminished secretion of OPG and an increased production of receptor activator of RANKL, as well as pro-inflammatory cytokines such as IL-1, IL-6, IL-11, and TNF-α. These alterations promote the formation and activation of osteoclasts, leading to increased bone resorption. The imbalance between osteoblast and osteoclast activity contributes to the acceleration of age-related bone loss, heightening the risk of osteoporosis and fractures. Furthermore, the decline in OPG levels facilitates increased interaction between RANKL and the receptor activator of nuclear factor κB (RANK), thereby amplifying osteoclastogenesis and bone resorption [47, 61]. Recent studies have demonstrated that the anabolic response to exercise is linked to the presence of Lamin A/C [62]. In humans, functional loading has been shown to enhance bone mass, with exercise training programs able to prevent or reverse about 1% of annual bone loss in the LS and femoral neck (FN) in both premenopausal and postmenopausal women. This finding highlights the significant role that mechanical loading, such as weight-bearing exercises, plays in maintaining bone health. Regular physical activity helps stimulate bone remodeling, promoting the maintenance of bone density and reducing the risk of osteoporosis and fractures. This effect highlights the importance of exercise in both preventing and mitigating age-related bone loss in women across various life stages [63, 64].
Bone mechanical behavior
The components of bone are precisely balanced to provide resistance to fractures while minimizing skeletal weight. Stiffness, which refers to the bone’s resistance to deformation, and strength, the maximum stress a bone can withstand before failure, are essential for supporting substantial loads and maintaining structural integrity. At the same time, toughness, or ductility, plays a critical role in absorbing impact energy, allowing bones to withstand forces without fracturing. This intricate balance between stiffness, strength, and toughness is crucial in ensuring that bones can both support mechanical demands and absorb stresses encountered during daily activities, thereby reducing the likelihood of injury. A shift in this balance toward higher tissue mineral content typically results in stiffer but more brittle bones.
Furthermore, changes in collagen structure can increase brittleness, as modifications in cross-linking not only stiffen the organic matrix but also alter the morphology of the mineral component, a topic explored further in this discussion. Notably, while BMD decreases in conditions such as osteoporosis, it increases in disorders like osteopetrosis [65-67]. BMD is widely regarded by researchers and clinicians as a key marker of fracture susceptibility, particularly due to its age-related decline in both men and women. Understanding the mechanisms behind the increased fragility and brittleness of bone with age is therefore essential. Numerous studies have demonstrated that cortical bone becomes progressively more brittle and weaker as individuals grow older [68-70].
Bone morphology
Morphology refers to the shape and structure of bones, which can be classified into several categories based on their form and function. These categories include long bones, such as the femur and tibia, which are primarily responsible for support and movement; short bones, like those found in the hands and feet, which provide stability and support with limited motion; and flat bones, such as the calvaria (skull) or sternum, which serve protective roles and provide a broad surface for muscle attachment. Each bone type is adapted to its specific functional requirements, contributing to the overall efficiency and resilience of the skeletal system. The morphological characteristics that influence bone strength include the size and shape of the bones [71-74].
During both development and aging, bone shape adapts in response to mechanical load, a process described by Wolff’s Law. According to this principle, bones remodel and adjust their structure in response to the stresses they experience, with increased load leading to bone formation and reduced load resulting in bone resorption. As bones undergo changes in shape—such as lengthening, thinning of their walls, or altering their center of gravity—their functional capabilities are also modified. These adaptations allow the skeletal system to optimize its strength and efficiency in response to changing mechanical demands, ensuring that bones remain resilient and capable of supporting the body’s activities throughout life [75-79]. A recent study examining the trabecular bones in the proximal tibias of 23-month-old and 5-month-old rats found a significant decrease in mineral density, bone volume fraction, and trabecular number in the older rats compared to the younger ones. These results underscore the impact of aging on bone structure, indicating that as rats age, there is a notable deterioration in critical bone characteristics that contribute to overall bone strength. The reduction in mineral density and trabecular number observed in the older rats reflects a decline in bone mass and structural integrity, which may increase the risk of fractures and other age-related bone pathologies [80].
In necropsied tibias from males aged 17 to 46 years, significant alterations in mechanical properties were found to correlate with the tibia’s size (slenderness), independent of age. As individuals aged, there was a noticeable increase in mechanical weakening, accompanied by more brittle bone behavior. These findings suggest that the structural changes in the tibia, particularly related to its size, contribute to the decline in mechanical strength over time, leading to increased fragility and a higher risk of fractures as age progresses [81].
During development, long bones undergo significant morphological changes. In the early stages of life, the cross-sectional geometry of these bones is characterized by a relatively uniform outer wall thickness. As the individual matures, this structure gradually transforms into a more ellipsoidal shape through a process known as “cortical drift.” This process involves remodeling the bone’s cortex, where the outer layer thickens and shifts, optimizing its shape and strength in response to mechanical forces. The resulting bone structure is more efficient in supporting the body’s weight and movement, adapting to the functional demands placed on the skeleton over time [82]. As with other transformations, bone geometry continuously changes throughout life in response to mechanical forces and biological signaling [82].
Bone cells
Bone is composed of several distinct cell types [83-86]. Osteoblastogenesis is regulated by various signaling pathways [87, 88]. Some osteoblasts differentiate into small, inactive bone-lining cells along dormant surfaces, while the remaining osteoblasts become embedded in mineralized matrix and develop long dendritic processes [88-90]. Connexin 43, the most prevalent gap junction protein in bone, plays a crucial role in bone modeling and remodeling [89, 90].
Osteoclast differentiation is reduced in older animals compared to younger ones, resulting in bone resorption rates that significantly exceed the rates of new bone formation. This imbalance between resorption and formation contributes to the progressive loss of bone mass and density, which is a hallmark of age-related bone degeneration. The impaired differentiation of osteoclasts in older animals may lead to an inefficient response to bone remodeling stimuli, further exacerbating the disparity between bone resorption and the ability to form new bone tissue [80, 91-93].
Osteoblasts, osteoclasts, and osteocytes have a limited lifespan, which is controlled by both intrinsic replication cycles and external influences. The Hayflick limit refers to the principle that cells can divide only a set number of times, with this replicative capacity gradually declining with age. This limitation is thought to be a result of telomere shortening during each cell division, leading to cellular senescence or apoptosis once the telomeres reach a critically short length. In bone cells, this reduction in proliferative capacity over time may contribute to decreased bone regeneration and impaired bone remodeling in older individuals [94]. The activity of telomerase is not infinite and decreases with age, resulting in a gradual reduction in the regenerative potential of stem cells over time [95]. Telomere damage induced by UV light and oxidative stress can hasten telomere shortening, thereby reducing cell lifespan. This heightened damage accelerates cellular aging and may play a significant role in the progressive decline of tissue regeneration and function over time [96].
Werner syndrome in humans is a genetic disorder that leads to the premature onset of age-related diseases, characterized by accelerated telomere shortening after puberty. This syndrome results from mutations in the WRN gene, which encodes a helicase involved in maintaining genomic stability. As a consequence, individuals with Werner syndrome experience accelerated cellular aging, with telomeres shortening at a significantly faster rate than in the general population. This rapid telomere attrition contributes to the early onset of conditions typically associated with aging, such as osteoporosis, cardiovascular diseases, and diabetes, underscoring the critical role of telomere maintenance in aging and age-related pathologies. This rapid telomere attrition contributes to the early development of aging-related conditions, including atherosclerosis, osteoporosis, and cancer [97, 98].
Cells often undergo changes in gene expression and activity with age, which may be attributed to alterations in gene (mRNA) translation. A study has demonstrated that treatments aimed at reducing mRNA translation in animals can extend their lifespan, implying that modulating protein synthesis could have an impact on aging and longevity [99]. Aging is particularly significant for bone cells, though it affects all cell types, as it is associated with a reduced ability to respond effectively to mechanical forces. This diminished responsiveness can negatively impact bone health and the regulation of bone remodeling, contributing to age-related skeletal problems [78].
Among the key cellular changes that occur with aging, contributing to the decline in bone’s mechanical function, are alterations in both the amount and rate of bone remodeling. Bone remodeling is a dynamic process that involves the resorption of old bone by osteoclasts, followed by the formation of new bone by osteoblasts. As individuals age, this process becomes less efficient, with a decrease in osteoblast activity and an increase in osteoclast activity. This imbalance results in a net loss of bone mass and changes in bone architecture, leading to decreased bone strength and increased fragility. Additionally, the reduced ability of osteoblasts to effectively regenerate bone tissue contributes to the overall decline in skeletal integrity, making bones more susceptible to fractures and other age-related conditions. With age, the amount of bone deposited during each remodeling cycle diminishes. This reduction may be attributed to a decline in the number of osteoblast precursors, a decrease in the stem cell population from which these precursors are derived, or a shortening of the lifespan of osteoblasts [100]. The signals that regulate the differentiation of osteoblast precursors decline with age, which likely contributes to the reduction in osteoblast numbers. These signaling pathways, including factors such as bone morphogenetic proteins (BMPs) and the Wnt/β-catenin signaling pathway, are essential for promoting the maturation of osteoblasts from their precursor cells. As these pathways become less effective with aging, the production of osteoblasts diminishes, leading to a decrease in bone formation. This reduction in osteoblast activity can contribute to the overall decline in bone mass and the increased risk of fractures typically observed in older individuals. This diminished signaling further impairs bone formation, leading to a decline in bone density and mechanical strength associated with aging [101]. In non-human primates, the number of hematopoietic cells, which serve as osteoclast precursors, decreases with age [100, 101].
Cell senescence may alter how cells respond to apoptotic signals, potentially influencing the way bone cells react to stress or damage. However, whether this occurs specifically in bone cells is an area that requires further investigation. Understanding the influence of senescence on bone cell function could offer valuable insights into age-related bone diseases and the mechanisms driving bone fragility [96].
Signaling pathways in bone cell aging and their impact on bone health
Various signaling pathways play a crucial role in regulating the differentiation and metabolism of bone cells, maintaining the delicate balance between bone formation and resorption. Among these, the Wnt/β-catenin pathway is essential for osteoblast differentiation and bone formation, while the RANK/RANKL/OPG signaling axis governs osteoclast differentiation and bone resorption. Additionally, BMPs are key regulators of osteoblast differentiation, and pathways such as Notch and Hedgehog further modulate bone cell activity. These interconnected pathways not only oversee the development of osteoblasts, osteoclasts, and osteocytes but also influence their function and survival, ensuring skeletal homeostasis. Disruptions in these signaling mechanisms can lead to bone-related disorders, such as osteoporosis, highlighting their importance in maintaining bone health [93, 102].
Signaling through a family of genes known as Wnt is thought to regulate cellular aging across various cell types. However, the exact mechanism by which Wnt signaling influences aging remains a topic of ongoing research and debate. The role of Wnt pathways in aging may involve regulating cellular processes such as proliferation, differentiation, and apoptosis, all of which are essential for maintaining bone homeostasis [103-106]. The findings suggest that sustaining or augmenting Wnt signaling may offer a potential approach to mitigate age-related bone loss and promote bone health in older individuals [107]. It suggests that increased levels of sFRP-4 may contribute to reduced bone mass and a higher risk of osteoporosis with aging, possibly by interfering with the regulation of osteoblast activity and bone formation [108].
Recent studies have suggested that oxidative stress may play a significant role in the aging of bone cells, with effects mediated indirectly through the Wnt signaling pathway. Reactive oxygen species are thought to activate FoxO transcription factors, which then bind to β-catenin. This interaction may disrupt the normal function of β-catenin, impairing osteoblast differentiation and function. As a result, oxidative stress may contribute to an imbalance in bone remodeling, potentially accelerating bone loss and increasing the risk of age-related bone diseases such as osteoporosis. This binding reduces the effective concentration of β-catenin in cells, subsequently impairing osteoblast formation. By disrupting the Wnt signaling pathway, oxidative stress hinders osteoblast differentiation and bone formation, thereby contributing to age-related bone loss [109, 110]. The reduction in Wnt signaling, triggered by PPAR-γ activation through ligands produced during lipid oxidation, also plays a role in the age-related decrease in osteoblast formation. This reduction in Wnt signaling impairs the differentiation and function of osteoblasts, thereby disrupting the balance between bone formation and resorption. As a result, the ability to regenerate bone tissue is diminished, leading to a decrease in bone mass and increased susceptibility to fractures with aging. This process exacerbates the bone cells’ inability to maintain bone mass and function with age. The activation of PPAR-γ, typically associated with adipogenesis, inhibits osteoblast differentiation, thereby disrupting the balance between bone resorption and formation. This imbalance results in weakened bone structure and an increased risk of fractures [65].
Bone protein
The organic matrix of bone is primarily composed of collagen, with type I collagen accounting for the majority, along with approximately 5% non-collagenous proteins [111]. Collagen cross-links exhibit distinct changes with aging, affecting the structural integrity and mechanical properties of the bone [112-114]. Building on this finding, the authors speculated that as aging progresses, the rate of nonenzymatic cross-linking increases, while the formation of mature enzymatic cross-links decreases. These alterations could reduce bone strength and toughness, ultimately compromising the bone’s ability to resist crack propagation. Similar findings have also been reported in bovine bone [115]. The accumulation of advanced glycation end products promotes osteoclast activity while inhibiting osteoblast differentiation, thereby contributing to the increased bone fragility associated with aging [116]. As tissue ages, the orientation of collagen fibers becomes more aligned, which can affect the mechanical properties of the bone [117]. With older age, changes occur in the expression and relative abundance of non-collagenous proteins in bone, which can impact its structural integrity and function [118, 119]. It is important to note that with age, not only do the distributions of non-collagenous proteins in bone change, but the extent of their post-translational modifications also diminishes [120, 121]. A decline in protein production with age has been documented, alongside an increase in bone matrix protein fragments in the older group [122].
Minerals
Bone mineral content, commonly known as “mineralization” or “ash content,” tends to increase with age. Classical research has shown that bone-breaking stress increases exponentially with higher ash content. However, once the ash content reaches its maximum level, bone toughness—the ability to resist fracture—begins to decline. This condition indicates that while greater mineralization strengthens the bone against breaking forces, excessive mineralization can lead to increased brittleness, thereby reducing the bone’s ability to absorb impact and making it more susceptible to fractures. This interplay between strength and toughness is essential for maintaining bone integrity over time [123].
The mineral present in bone is a form of hydroxyapatite, a naturally occurring mineral with the chemical formula [Ca10(PO4)6(OH)2]. Bone mineral crystals are nanoscale structures, typically measuring about 1 to 1.5 nm in thickness, 5 to 25 nm in width, and 8 to 40 nm in length. These nanoscale dimensions enable the unique mechanical properties of bone, including its strength and flexibility, which contribute to its ability to support and protect the body while maintaining a light and resilient structure [124-126]. These bone mineral crystals also incorporate various inclusions and substitutions that evolve with age. A notable substitution is carbonate, which replaces hydroxyl and phosphate groups within the surface and crystal lattice of apatite. This alteration can impact the structural characteristics of the crystals, potentially modifying their mechanical properties. As aging progresses, these changes in the mineral composition may affect bone mineralization, contributing to the decline in bone strength and an increased risk of fractures [127-129].
Conclusion
The aging process significantly disrupts bone remodeling, leading to a decline in bone mass, strength, and structural integrity. Key factors, including hormonal imbalances, elevated PTH levels, and the accumulation of bone marrow fat, contribute to the deterioration of bone health. Additionally, changes in bone cell functionality and alterations in the bone matrix and mineral composition further compromise bone strength and toughness. As these changes accelerate the risk of osteoporosis and fractures, understanding the molecular and cellular mechanisms underlying bone aging is crucial for developing effective therapeutic approaches. Continued research is essential for identifying novel interventions that can mitigate the adverse effects of aging on bone health and enhance the quality of life in the aging population.
Ethical Considerations
Compliance with ethical guidelines
There were no ethical considerations to be considered in this research.
Funding
This study was supported by the Iran University of Medical Sciences, Tehran, Iran.
Authors' contributions
Conceptualization and supervision: Saeedreza Amiri; Methodology: Pouria Tabrizian, and Morteza Nakhaei Amroodi; Data collection: Khatere Mokhtari, and Mohammadreza Bahaeddini; Data analysis: Khatere Mokhtari and Mohammadreza Bahaeddini; Investigation and writing: All authors; Funding acquisition and Resources: Saeedreza Amiri.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors wish to thank all individuals who helped us develop this research.
References
- Raisz LG, Rodan GA. Pathogenesis of osteoporosis. Endocrinol Metab Clin North Am. 2003 ;32(1):15-24. [DOI:10.1016/S0889-8529(02)00055-5] [PMID]
- Baniasadi M, Talebi S, Mokhtari K, Zabolian AH, Khosroshahi EM, Entezari M, et al. Role of non-coding RNAs in osteoporosis. Pathol Res Pract. 2024; 253:155036. [DOI:10.1016/j.prp.2023.155036]
- Riggs BL, Khosla S, Melton LJ 3rd. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002; 23(3):279-302. [DOI:10.1210/edrv.23.3.0465] [PMID]
- Raisz LG, Seeman E. Causes of age-related bone loss and bone fragility: An alternative view. J Bone Miner Res. 2001; 16(11):1948-52. [DOI:10.1359/jbmr.2001.16.11.1948] [PMID]
- Slemenda C, Longcope C, Peacock M, Hui S, Johnston CC. Sex steroids, bone mass, and bone loss. A prospective study of pre-, peri-, and postmenopausal women. J Clin Invest. 1996; 97(1):14-21. [DOI:10.1172/JCI118382] [PMID]
- Rosen CJ, Donahue LR, Hunter SJ. Insulin-like growth factors and bone: The osteoporosis connection. Proc Soc Exp Biol Med. 1994; 206(2):83-102. [DOI:10.3181/00379727-206-43726] [PMID]
- Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin North Am. 2005; 34(4):1015-30, xi. [DOI:10.1016/j.ecl.2005.07.009]
- Riggs BL, Melton Iii LJ 3rd, Robb RA, Camp JJ, Atkinson EJ, et al. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. J Bone Miner Res. 2004; 19(12):1945-54. [DOI:10.1359/jbmr.040916] [PMID]
- Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: Consequences for bone loss and fractures and therapeutic implications. Endocr Rev. 2001; 22(4):477-501. [DOI:10.1210/edrv.22.4.0437]
- Ledger GA, Burritt MF, Kao PC, O'Fallon WM, Riggs BL, Khosla S. Role of parathyroid hormone in mediating nocturnal and age-related increases in bone resorption. J Clin Endocrinol Metab. 1995; 80(11):3304-10. [PMID]
- Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008; 29(4):441-64. [DOI:10.1210/er.2008-0002]
- Khosla S. Update in male osteoporosis. J Clin Endocrinol Metab. 2010; 95(1):3-10. [DOI:10.1210/jc.2009-1740] [PMID]
- Kennel KA, Riggs BL, Achenbach SJ, Oberg AL, Khosla S. Role of parathyroid hormone in mediating age-related changes in bone resorption in men. Osteoporos Int. 2003; 14(8):631-6. [DOI:10.1007/s00198-003-1417-0] [PMID]
- Khosla S, Atkinson EJ, Melton LJ 3rd, Riggs BL. Effects of age and estrogen status on serum parathyroid hormone levels and biochemical markers of bone turnover in women: A population-based study. J Clin Endocrinol Metab. 1997; 82(5):1522-7. [DOI:10.1210/jcem.82.5.3946]
- Khosla S, Riggs BL, Robb RA, Camp JJ, Achenbach SJ, Oberg AL, et al. Relationship of volumetric bone density and structural parameters at different skeletal sites to sex steroid levels in women. J Clin Endocrinol Metab. 2005; 90(9):5096-103. [DOI:10.1210/jc.2005-0396] [PMID]
- McCauley LK, Tözüm TF, Kozloff KM, Koh-Paige AJ, Chen C, Demashkieh M, et al. Transgenic models of metabolic bone disease: Impact of estrogen receptor deficiency on skeletal metabolism. Connect Tissue Res. 2003; 44 (Suppl 1):250-63. [DOI:10.1080/03008200390181744]
- Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999; 140(9):4367-70. [DOI:10.1210/endo.140.9.7131] [PMID]
- Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996; 2(10):1132-6. [DOI:10.1038/nm1096-1132]
- Lundberg P, Lundgren I, Mukohyama H, Lehenkari PP, Horton MA, Lerner UH. Vasoactive intestinal peptide (VIP)/pituitary adenylate cyclase-activating peptide receptor subtypes in mouse calvarial osteoblasts: Presence of VIP-2 receptors and differentiation-induced expression of VIP-1 receptors. Endocrinology. 2001; 142(1):339-47. [DOI:10.1210/endo.142.1.7912] [PMID]
- Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003; 111(8):1221-30. [DOI:10.1172/JCI200317215] [PMID]
- Lundberg P, Boström I, Mukohyama H, Bjurholm A, Smans K, Lerner UH. Neuro-hormonal control of bone metabolism: Vasoactive intestinal peptide stimulates alkaline phosphatase activity and mRNA expression in mouse calvarial osteoblasts as well as calcium accumulation mineralized bone nodules. Regul Pept. 1999; 85(1):47-58. [DOI:10.1016/S0167-0115(99)00069-5]
- Mitnick MA, Grey A, Masiukiewicz U, Bartkiewicz M, Rios-Velez L, Friedman S, et al. Parathyroid hormone induces hepatic production of bioactive interleukin-6 and its soluble receptor. Am J Physiol Endocrinol Metab. 2001; 280(3):E405-12. [DOI:10.1152/ajpendo.2001.280.3.E405] [PMID]
- Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of blockade of TNF-alpha and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res. 2007; 22(5):724-9. [DOI:10.1359/jbmr.070207] [PMID]
- Oursler MJ, Pederson L, Fitzpatrick L, Riggs BL, Spelsberg T. Human giant cell tumors of the bone (osteoclastomas) are estrogen target cells. Proc Natl Acad Sci U S A. 1994; 91(12):5227-31. [DOI:10.1073/pnas.91.12.5227]
- Perrien DS, Achenbach SJ, Bledsoe SE, Walser B, Suva LJ, Khosla S, et al. Bone turnover across the menopause transition: Correlations with inhibins and follicle-stimulating hormone. J Clin Endocrinol Metab. 2006; 91(5):1848-54. [DOI:10.1210/jc.2005-2423] [PMID]
- Slemenda CW, Longcope C, Zhou L, Hui SL, Peacock M, Johnston CC. Sex steroids and bone mass in older men. Positive associations with serum estrogens and negative associations with androgens. J Clin Invest. 1997; 100(7):1755-9. [DOI:10.1172/JCI119701] [PMID]
- Khosla S, Melton LJ 3rd, Atkinson EJ, O'Fallon WM. Relationship of serum sex steroid levels to longitudinal changes in bone density in young versus elderly men. J Clin Endocrinol Metab. 2001; 86(8):3555-61. [DOI:10.1210/jcem.86.8.7736]
- Mellström D, Vandenput L, Mallmin H, Holmberg AH, Lorentzon M, Odén A, et al. Older men with low serum estradiol and high serum SHBG have an increased risk of fractures. J Bone Miner Res. 2008; 23(10):1552-60. [DOI:10.1359/jbmr.080518]
- Szulc P, Munoz F, Claustrat B, Garnero P, Marchand F, Duboeuf F, et al. Bioavailable estradiol may be an important determinant of osteoporosis in men: The MINOS study. J Clin Endocrinol Metab. 2001; 86(1):192-9. [PMID]
- Falahati-Nini A, Riggs BL, Atkinson EJ, O'Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest. 2000; 106(12):1553-60. [DOI:10.1172/JCI10942]
- Leder BZ, LeBlanc KM, Schoenfeld DA, Eastell R, Finkelstein JS. Differential effects of androgens and estrogens on bone turnover in normal men. J Clin Endocrinol Metab. 2003; 88(1):204-10. [DOI:10.1210/jc.2002-021036] [PMID]
- Sanyal A, Hoey KA, Mödder UI, Lamsam JL, McCready LK, Peterson JM, et al. Regulation of bone turnover by sex steroids in men. J Bone Miner Res. 2008; 23(5):705-14. [DOI:10.1359/jbmr.071212] [PMID]
- LeBlanc ES, Nielson CM, Marshall LM, Lapidus JA, Barrett-Connor E, Ensrud KE, et al. The effects of serum testosterone, estradiol, and sex hormone binding globulin levels on fracture risk in older men. J Clin Endocrinol Metab. 2009; 94(9):3337-46. [DOI:10.1210/jc.2009-0206] [PMID]
- Clarke BL, Khosla S. Physiology of bone loss. Radiol Clin North Am. 2010; 48(3):483-95. [DOI:10.1016/j.rcl.2010.02.014]
- Rosen CJ, Bouxsein ML. Mechanisms of disease: Is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006; 2(1):35-43. [DOI:10.1038/ncprheum0070] [PMID]
- Perrien DS, Akel NS, Dupont-Versteegden EE, Skinner RA, Siegel ER, Suva LJ, et al. Aging alters the skeletal response to disuse in the rat. Am J Physiol Regul Integr Comp Physiol. 2007; 292(2):R988-96. [DOI:10.1152/ajpregu.00302.2006] [PMID]
- Verma S, Rajaratnam JH, Denton J, Hoyland JA, Byers RJ. Adipocytic proportion of bone marrow is inversely related to bone formation in osteoporosis. J Clin Pathol. 2002; 55(9):693-8. [DOI:10.1136/jcp.55.9.693]
- Meunier P, Aaron J, Edouard C, Vignon G. Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin Orthop Relat Res. 1971; 80:147-54. [DOI:10.1097/00003086-197110000-00021] [PMID]
- Duque G, Rivas D, Li W, Li A, Henderson JE, Ferland G, et al. Age-related bone loss in the LOU/c rat model of healthy ageing. Exp Gerontol. 2009; 44(3):183-9. [DOI:10.1016/j.exger.2008.10.004] [PMID]
- Griffith JF, Yeung DK, Antonio GE, Lee FK, Hong AW, Wong SY, et al. Vertebral bone mineral density, marrow perfusion, and fat content in healthy men and men with osteoporosis: dynamic contrast-enhanced MR imaging and MR spectroscopy. Radiology. 2005; 236(3):945-51. [DOI:10.1148/radiol.2363041425] [PMID]
- Shen W, Chen J, Punyanitya M, Shapses S, Heshka S, Heymsfield SB. MRI-measured bone marrow adipose tissue is inversely related to DXA-measured bone mineral in Caucasian women. Osteoporos Int. 2007; 18(5):641-7. [DOI:10.1007/s00198-006-0285-9]
- Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001; 2(3):165-71. [DOI:10.1023/A:1011513223894] [PMID]
- Rosen CJ, Ackert-Bicknell C, Rodriguez JP, Pino AM. Marrow fat and the bone microenvironment: Developmental, functional, and pathological implications. Crit Rev Eukaryot Gene Expr. 2009; 19(2):109-24. [DOI:10.1615/CritRevEukarGeneExpr.v19.i2.20] [PMID]
- Zhou S, Greenberger JS, Epperly MW, Goff JP, Adler C, Leboff MS, et al. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell. 2008; 7(3):335-43. [DOI:10.1111/j.1474-9726.2008.00377.x]
- Gimble JM, Zvonic S, Floyd ZE, Kassem M, Nuttall ME. Playing with bone and fat. J Cell Biochem. 2006; 98(2):251-66. [DOI:10.1002/jcb.20777] [PMID]
- Lecka-Czernik B. PPARs and bone metabolism. PPAR Res. 2006; 2006:18089. [DOI:10.1155/PPAR/2006/18089]
- Duque G, Troen BR. Understanding the mechanisms of senile osteoporosis: New facts for a major geriatric syndrome. J Am Geriatr Soc. 2008; 56(5):935-41. [DOI:10.1111/j.1532-5415.2008.01764.x] [PMID]
- Maurin AC, Chavassieux PM, Frappart L, Delmas PD, Serre CM, Meunier PJ. Influence of mature adipocytes on osteoblast proliferation in human primary cocultures. Bone. 2000; 26(5):485-9. [DOI:10.1016/S8756-3282(00)00252-0] [PMID]
- Musacchio E, Priante G, Budakovic A, Baggio B. Effects of unsaturated free fatty acids on adhesion and on gene expression of extracellular matrix macromolecules in human osteoblast-like cell cultures. Connect Tissue Res. 2007; 48(1):34-8. [DOI:10.1080/03008200601056528] [PMID]
- Felson DT, Zhang Y, Hannan MT, Anderson JJ. Effects of weight and body mass index on bone mineral density in men and women: The Framingham study. J Bone Miner Res. 1993; 8(5):567-73. [DOI:10.1002/jbmr.5650080507] [PMID]
- Lindsay R, Cosman F, Herrington BS, Himmelstein S. Bone mass and body composition in normal women. J Bone Miner Res. 1992; 7(1):55-63. [DOI:10.1002/jbmr.5650070109]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996; 334(5):292-5. [DOI:10.1056/NEJM199602013340503] [PMID]
- Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999; 140(4):1630-8. [DOI:10.1210/endo.140.4.6637]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell. 2000; 100(2):197-207. [DOI:10.1016/S0092-8674(00)81558-5]
- Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002; 111(3):305-17. [DOI:10.1016/S0092-8674(02)01049-8] [PMID]
- Shi Y, Yadav VK, Suda N, Liu XS, Guo XE, Myers MG Jr, et al. Dissociation of the neuronal regulation of bone mass and energy metabolism by leptin in vivo. Proc Natl Acad Sci U S A. 2008; 105(51):20529-33. [DOI:10.1073/pnas.0808701106]
- Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C, et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009; 138(5):976-89. [DOI:10.1016/j.cell.2009.06.051] [PMID]
- Mödder UI, Achenbach SJ, Amin S, Riggs BL, Melton LJ 3rd, Khosla S. Relation of serum serotonin levels to bone density and structural parameters in women. J Bone Miner Res. 2010; 25(2):415-22. [DOI:10.1359/jbmr.090721] [PMID]
- Seeman E. From density to structure: Growing up and growing old on the surfaces of bone. J Bone Miner Res. 1997; 12(4):509-21. [DOI:10.1359/jbmr.1997.12.4.509] [PMID]
- Riggs BL, Melton LJ 3rd. Involutional osteoporosis. N Engl J Med. 1986; 314(26):1676-86. [DOI:10.1056/NEJM198606263142605]
- Weinreb M, Rodan GA, Thompson DD. Osteopenia in the immobilized rat hind limb is associated with increased bone resorption and decreased bone formation. Bone. 1989; 10(3):187-94. [DOI:10.1016/8756-3282(89)90052-5]
- Duque G, Li W, Yeo LS, Vidal C, Fatkin D. Attenuated anabolic response to exercise in lamin A/C haploinsufficient mice. Bone. 2011; 49(3):412-8. [DOI:10.1016/j.bone.2011.04.023] [PMID]
- Leichter I, Simkin A, Margulies JY, Bivas A, Steinberg R, Giladi M, et al. Gain in mass density of bone following strenuous physical activity. J Orthop Res. 1989; 7(1):86-90. [DOI:10.1002/jor.1100070112] [PMID]
- Wolff I, van Croonenborg JJ, Kemper HC, Kostense PJ, Twisk JW. The effect of exercise training programs on bone mass: a meta-analysis of published controlled trials in pre- and postmenopausal women. Osteoporos Int. 1999; 9(1):1-12. [DOI:10.1007/s001980050109] [PMID]
- Manolagas SC. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010; 31(3):266-300. [DOI:10.1210/er.2009-0024] [PMID]
- Kaste SC, Kasow KA, Horwitz EM. Quantitative bone mineral density assessment in malignant infantile osteopetrosis. Pediatr Blood Cancer. 2007; 48(2):181-5. [DOI:10.1002/pbc.20759]
- Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet. 2002; 359(9321):1929-36. [DOI:10.1016/S0140-6736(02)08761-5] [PMID]
- Currey J, Butler G. The mechanical properties of bone tissue in children. J Bone Joint Surg Am. 1975; 57(6):810-4. [DOI:10.2106/00004623-197557060-00015]
- Burstein AH, Reilly DT, Martens M. Aging of bone tissue: Mechanical properties. J Bone Joint Surg Am. 1976; 58(1):82-6. [DOI:10.2106/00004623-197658010-00015] [PMID]
- Tommasini SM, Nasser P, Jepsen KJ. Sexual dimorphism affects tibia size and shape but not tissue-level mechanical properties. Bone. 2007; 40(2):498-505. [DOI:10.1016/j.bone.2006.08.012] [PMID]
- Jepsen KJ. Systems analysis of bone. Wiley Interdiscip Rev Syst Biol Med. 2009; 1(1):73-88. [DOI:10.1002/wsbm.15]
- Seeman E. The structural and biomechanical basis of the gain and loss of bone strength in women and men. Endocrinol Metab Clin North Am. 2003; 32(1):25-38. [DOI:10.1016/S0889-8529(02)00078-6] [PMID]
- Wang Q, Seeman E. Skeletal growth and peak bone strength. Best Pract Res Clin Endocrinol Metab. 2008; 22(5):687-700. [DOI:10.1016/j.beem.2008.07.008] [PMID]
- Seeman E. Bone modeling and remodeling. Crit Rev Eukaryot Gene Expr. 2009; 19(3):219-33. [DOI:10.1615/CritRevEukarGeneExpr.v19.i3.40]
- Ramanadham S, Yarasheski KE, Silva MJ, Wohltmann M, Novack DV, Christiansen B, et al. Age-related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2beta)-null mice. Am J Pathol. 2008; 172(4):868-81. [DOI:10.2353/ajpath.2008.070756] [PMID]
- Woo SL, Kuei SC, Amiel D, Gomez MA, Hayes WC, White FC, et al. The effect of prolonged physical training on the properties of long bone: A study of Wolff's Law. J Bone Joint Surg Am. 1981; 63(5):780-7. [DOI:10.2106/00004623-198163050-00013]
- Wallace JM, Rajachar RM, Allen MR, Bloomfield SA, Robey PG, Young MF, et al. Exercise-induced changes in the cortical bone of growing mice are bone- and gender-specific. Bone. 2007; 40(4):1120-7. [DOI:10.1016/j.bone.2006.12.002] [PMID]
- Chen JH, Liu C, You L, Simmons CA. Boning up on Wolff's Law: Mechanical regulation of the cells that make and maintain bone. J Biomech. 2010; 43(1):108-18. [DOI:10.1016/j.jbiomech.2009.09.016] [PMID]
- Kawashima Y, Fritton JC, Yakar S, Epstein S, Schaffler MB, Jepsen KJ, et al. Type 2 diabetic mice demonstrate slender long bones with increased fragility secondary to increased osteoclastogenesis. Bone. 2009; 44(4):648-55. [DOI:10.1016/j.bone.2008.12.012]
- Pietschmann P, Skalicky M, Kneissel M, Rauner M, Hofbauer G, Stupphann D, et al. Bone structure and metabolism in a rodent model of male senile osteoporosis. Exp Gerontol. 2007; 42(11):1099-108. [DOI:10.1016/j.exger.2007.08.008] [PMID]
- Tommasini SM, Nasser P, Schaffler MB, Jepsen KJ. Relationship between bone morphology and bone quality in male tibias: Implications for stress fracture risk. J Bone Miner Res. 2005;2 0(8):1372-80. [DOI:10.1359/JBMR.050326]
- Goldman HM, McFarlin SC, Cooper DM, Thomas CD, Clement JG. Ontogenetic patterning of cortical bone microstructure and geometry at the human mid-shaft femur. Anat Rec (Hoboken). 2009; 292(1):48-64. [DOI:10.1002/ar.20778] [PMID]
- WHO. Assessment of fracture risk and its application to screening for postmenopausal oseteoporosis. WHO technical report series. Geneva: WHO;1994. [Link]
- Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008; 42(4):606-15. [DOI:10.1016/j.bone.2007.12.224] [PMID]
- Boyce BF, Yao Z, Xing L. Osteoclasts have multiple roles in bone in addition to bone resorption. Crit Rev Eukaryot Gene Expr. 2009; 19(3):171-80. [DOI:10.1615/CritRevEukarGeneExpr.v19.i3.10] [PMID]
- Adachi T, Aonuma Y, Ito S, Tanaka M, Hojo M, Takano-Yamamoto T, et al. Osteocyte calcium signaling response to bone matrix deformation. J Biomech. 2009; 42(15):2507-12. [DOI:10.1016/j.jbiomech.2009.07.006] [PMID]
- Jilka RL, Weinstein RS, Parfitt AM, Manolagas SC. Quantifying osteoblast and osteocyte apoptosis: Challenges and rewards. J Bone Miner Res. 2007; 22(10):1492-501. [DOI:10.1359/jbmr.070518]
- Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. 2010; 21(6):369-74. [DOI:10.1016/j.tem.2010.01.010] [PMID]
- Matsuo K. Cross-talk among bone cells. Curr Opin Nephrol Hypertens. 2009; 18(4):292-7. [DOI:10.1097/MNH.0b013e32832b75f1] [PMID]
- Rochefort GY, Pallu S, Benhamou C-L. Osteocyte: The unrecognized side of bone tissue. Osteoporos Int. 2010; 21(9):1457-69. [DOI:10.1007/s00198-010-1194-5] [PMID]
- Cao JJ, Wronski TJ, Iwaniec U, Phleger L, Kurimoto P, Boudignon B, et al. Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J Bone Miner Res. 2005; 20(9):1659-68. [DOI:10.1359/JBMR.050503] [PMID]
- Henriksen K, Leeming DJ, Byrjalsen I, Nielsen RH, Sorensen MG, Dziegiel MH, et al. Osteoclasts prefer aged bone. Osteoporos Int. 2007; 18(6):751-9. [DOI:10.1007/s00198-006-0298-4] [PMID]
- Ginaldi L, Mengoli LP, De Martinis M. Osteoporosis, inflammation and ageing. In: Fulop T, Franceschi C, Hirokawa K, Pawelec G, editors. Handbook on Immunosenescence. Dordrecht: Springer; 2009. [DOI:10.1007/978-1-4020-9063-9_64]
- Martin G, Sprague CA, Epstein CJ. Replicative lifespan of cultivated human cells. Effects of donor's age, tissue, and genotype. Lab Invest. 1970; 23(1):86-92. [PMID]
- Calado RT. Telomeres and marrow failure. Hematology Am Soc Hematol Educ Program. 2009; 338-43. [DOI:10.1182/asheducation-2009.1.338]
- Muller M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal. 2009; 11(1):59-98. [DOI:10.1089/ars.2008.2104] [PMID]
- Martin GM. Genetic modulation of senescent phenotypes in Homo sapiens. Cell. 2005; 120(4):523-32. [DOI:10.1016/j.cell.2005.01.031] [PMID]
- Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004; 36(8):877-82. [DOI:10.1038/ng1389] [PMID]
- Kennedy BK, Kaeberlein M. Hot topics in aging research: Protein translation, 2009. Aging Cell. 2009; 8(6):617-23. [DOI:10.1111/j.1474-9726.2009.00522.x] [PMID]
- Szulc P, Seeman E. Thinking inside and outside the envelopes of bone: Dedicated to PDD. Osteoporos Int. 2009; 20(8):1281-8. [DOI:10.1007/s00198-009-0994-y]
- Lee CC, Fletcher MD, Tarantal AF. Effect of age on the frequency, cell cycle, and lineage maturation of rhesus monkey (Macaca mulatta) CD34+ and hematopoietic progenitor cells. Pediatr Res. 2005; 58(2):315-22. [DOI:10.1203/01.PDR.0000169975.30339.32] [PMID]
- Chau JF, Leong WF, Li B. Signaling pathways governing osteoblast proliferation, differentiation and function. Histol Histopathol. 2009; 24(12):1593-606. [DOI: 10.14670/HH-24.1593] [PMID]
- Williams BO, Insogna KL. Where Wnts went: The exploding field of Lrp5 and Lrp6 signaling in bone. J Bone Miner Res. 2009; 24(2):171-8. [DOI:10.1359/jbmr.081235] [PMID]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007; 317(5839):807-10. [DOI:10.1126/science.1144090]
- DeCarolis NA, Wharton Jr KA, Eisch AJ. Which way does the Wnt blow? Exploring the duality of canonical Wnt signaling on cellular aging. Bioessays. 2008; 30(2):102-6. [DOI:10.1002/bies.20709] [PMID]
- Rauner M, Sipos W, Pietschmann P. Age-dependent Wnt gene expression in bone and during the course of osteoblast differentiation. Age (Dordr). 2008; 30(4):273-82. [DOI:10.1007/s11357-008-9069-9] [PMID]
- Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005; 102(9):3324-9. [DOI:10.1073/pnas.0408742102] [PMID]
- Nakanishi R, Shimizu M, Mori M, Akiyama H, Okudaira S, Otsuki B, et al. Secreted frizzled-related protein 4 is a negative regulator of peak BMD in SAMP6 mice. J Bone Miner Res. 2006; 21(11):1713-21. [DOI:10.1359/jbmr.060719] [PMID]
- Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008; 20(2):126-36. [DOI:10.1016/j.ceb.2008.02.005] [PMID]
- Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007; 282(37):27285-97. [DOI:10.1074/jbc.M702810200] [PMID]
- Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: A possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010; 21(2):195-214. [DOI:10.1007/s00198-009-1066-z]
- Vashishth D, Gibson G, Khoury J, Schaffler M, Kimura J, Fyhrie DP. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone. 2001; 28(2):195-201. [DOI:10.1016/S8756-3282(00)00434-8] [PMID]
- Boskey AL, Coleman R. Aging and bone. J Dent Res. 2010; 89(12):1333-48. [DOI:10.1177/0022034510377791] [PMID]
- Banse X, Devogelaer JP, Lafosse A, Sims TJ, Grynpas M, Bailey AJ. Cross-link profile of bone collagen correlates with structural organization of trabeculae. Bone. 2002; 31(1):70-6. [DOI:10.1016/S8756-3282(02)00800-1] [PMID]
- Tang S, Zeenath U, Vashishth D. Effects of nonenzymatic glycation on cancellous bone fragility. Bone. 2007; 40(4):1144-51. [DOI:10.1016/j.bone.2006.12.056]
- Viguet-Carrin S, Follet H, Gineyts E, Roux JP, Munoz F, Chapurlat R, et al. Association between collagen cross-links and trabecular microarchitecture properties of human vertebral bone. Bone. 2010; 46(2):342-7. [DOI:10.1016/j.bone.2009.10.001] [PMID]
- Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol. 2003; 21(11):1356-60. [DOI:10.1038/nbt894] [PMID]
- Ikeda T, Nagai Y, Yamaguchi A, Yokose S, Yoshiki S. Age-related reduction in bone matrix protein mRNA expression in rat bone tissues: Application of histomorphometry to in situ hybridization. Bone. 1995; 16(1):17-23. [DOI:10.1016/s8756-3282(94)00002-6] [PMID]
- Zhu K, Prince RL. Lifestyle and osteoporosis. Curr Osteoporos Rep. 2015; 13(1):52-9. [DOI:10.1007/s11914-014-0248-6] [PMID]
- Plantalech L, Guillaumont M, Vergnaud P, Leclercq M, Delmas PD. Impairment of gamma carboxylation of circulating osteocalcin (bone gla protein) in elderly women. J Bone Miner Res. 1991; 6(11):1211-6. [DOI:10.1002/jbmr.5650061111] [PMID]
- Grzesik WJ, Frazier CR, Shapiro JR, Sponseller PD, Robey PG, Fedarko NS. Age-related changes in human bone proteoglycan structure. Impact of osteogenesis imperfecta. J Biol Chem. 2002; 277(46):43638-47. [DOI:10.1074/jbc.M202124200] [PMID]
- Grynpas M, Tupy J, Sodek J. The distribution of soluble, mineral-bound, and matrix-bound proteins in osteoporotic and normal bones. Bone. 1994; 15(5):505-13. [DOI:10.1016/8756-3282(94)90274-7]
- Currey JD. The relationship between the stiffness and the mineral content of bone. J Biomech. 1969; 2(4):477-80. [DOI:10.1016/0021-9290(69)90023-2] [PMID]
- Eppell SJ, Tong W, Katz JL, Kuhn L, Glimcher MJ. Shape and size of isolated bone mineralites measured using atomic force microscopy. J Orthop Res. 2001; 19(6):1027-34. [DOI:10.1016/S0736-0266(01)00034-1]
- Tong W, Glimcher MJ, Katz JL, Kuhn L, Eppell SJ. Size and shape of mineralites in young bovine bone measured by atomic force microscopy. Calcif Tissue Int. 2003; 72(5):592-8 [DOI:10.1007/s00223-002-1077-7] [PMID]
- Burger C, Zhou HW, Wang H, Sics I, Hsiao BS, Chu B, et al. Lateral packing of mineral crystals in bone collagen fibrils. Biophys J. 2008; 95(4):1985-92. [DOI:10.1529/biophysj.107.128355]
- LeGeros RZ. Properties of osteoconductive biomaterials: calcium phosphates. Clin Orthop Relat Res. 2002; (395):81-98. [DOI:10.1097/00003086-200202000-00009] [PMID]
- Handschin R, Stern W. X-ray diffraction studies on the lattice perfection of human bone apatite (Crista iliaca). Bone. 1995; 16(4):S355-S63. [DOI:10.1016/S8756-3282(95)80385-8] [PMID]
- Meneghini C, Dalconi MC, Nuzzo S, Mobilio S, Wenk RH. Rietveld refinement on X-ray diffraction patterns of bioapatite in human fetal bones. Biophys J. 2003; 84(3):2021-9. [DOI:10.1016/S0006-3495(03)75010-3] [PMID]
Type of Study: Review Paper |
Subject:
Trauma
Received: 2025/03/9 | Accepted: 2025/03/15 | Published: 2024/05/31
Received: 2025/03/9 | Accepted: 2025/03/15 | Published: 2024/05/31
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |

Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
