

Volume 12, Issue 2 (Spring 2025)
JROS 2025, 12(2): 91-96 |
Back to browse issues page



Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Kargar Shooroki K, Hajialiloo Sami S, HamidzadehKhiavi A, Sour B, Movahedi M S, Roshanravan B. Extraskeletal Ewing’s Sarcoma of the Foot in a Pediatric Patient: A Case Report. JROS 2025; 12 (2) :91-96
URL: http://jros.iums.ac.ir/article-1-2305-en.html
URL: http://jros.iums.ac.ir/article-1-2305-en.html
Khalil Kargar Shooroki1 

 , Sam Hajialiloo Sami1 , Amin HamidzadehKhiavi2 , Behnam Sour1 , Mohammad Saleh Movahedi1 , Babak Roshanravan2
, Sam Hajialiloo Sami1 , Amin HamidzadehKhiavi2 , Behnam Sour1 , Mohammad Saleh Movahedi1 , Babak Roshanravan2
, Sam Hajialiloo Sami1 , Amin HamidzadehKhiavi2 , Behnam Sour1 , Mohammad Saleh Movahedi1 , Babak Roshanravan2
1- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran.
2- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran. & Department of Orthopedic Surgery, School of Medicine, Imam Reza Hospital, Birjand University of Medical Sciences, Birjand, Iran.
2- Department of Orthopedics, Bone and Joint Reconstruction Research Center, School of Medicine, Iran University of Medical Sciences, Tehran, Iran. & Department of Orthopedic Surgery, School of Medicine, Imam Reza Hospital, Birjand University of Medical Sciences, Birjand, Iran.
Full-Text [PDF 1809 kb]
(83 Downloads)
| Abstract (HTML) (212 Views)
Full-Text: (47 Views)
Introduction
Ewing sarcoma (ES), described by J. Ewing in 1921, is the second most common type of primary malignant bone tumor in children and young individuals. Although ES is typically considered a pediatric tumor, it can impact individuals up to the age of thirty [1]. It has been reported that about 25% of these aggressive tumors originate from soft tissue, known as extraskeletal ES (EES) [2]. EES, first documented in 1969 by Tefft et al., is an extremely uncommon entity that typically manifests in pediatrics and adolescents. Its incidence is reported to be about 1 in 5–10 million people, with most cases ranging between 10 and 20 years old [3, 4].
EES belongs to the ES family of tumors (ESFT), and its spherical malignant cells resemble one another in their molecular lineage [2, 5, 6]. According to available data, EES usually originates from the chest wall, proximal thigh, and extremities [7]. However, involvement of the breast, liver, and urinary tract has also been documented [8, 9]. The typical clinical manifestation of EES is an expanding lesion of about 2–3 cm, inducing compression symptoms, usually focal pain [7]. The mass is usually soft, movable, and superficial. Imaging studies, most importantly (MRI), are usually the first step in diagnosis, confirmed by histology and immunohistochemistry evaluations. Surgical resection, chemotherapy, and radiotherapy (in unresectable cases) are treatment options for EES management [2, 10].
Knowledge regarding the clinical features and incidence of EES in pediatric patients is limited. This study aimed to report an unusual case of EES of the foot in a 4-year-old Iranian boy, who was admitted with pain and a wart-like lesion on the plantar surface of his left foot in the first web space for the past 4 months. The diagnosis of subcutaneous EES in the foot is rare, with only a few cases reported at such a young age [11]. Although it is an uncommon site, it is important to consider EES, especially if there is a visible cutaneous lesion. The rarity of EES, along with relatively nonspecific findings in histology and immunophenotype, makes the diagnosis of superficial EES challenging, and radiologists, pathologists, and, more importantly, clinicians should consider these tumors in their differential diagnosis.
Case Presentation
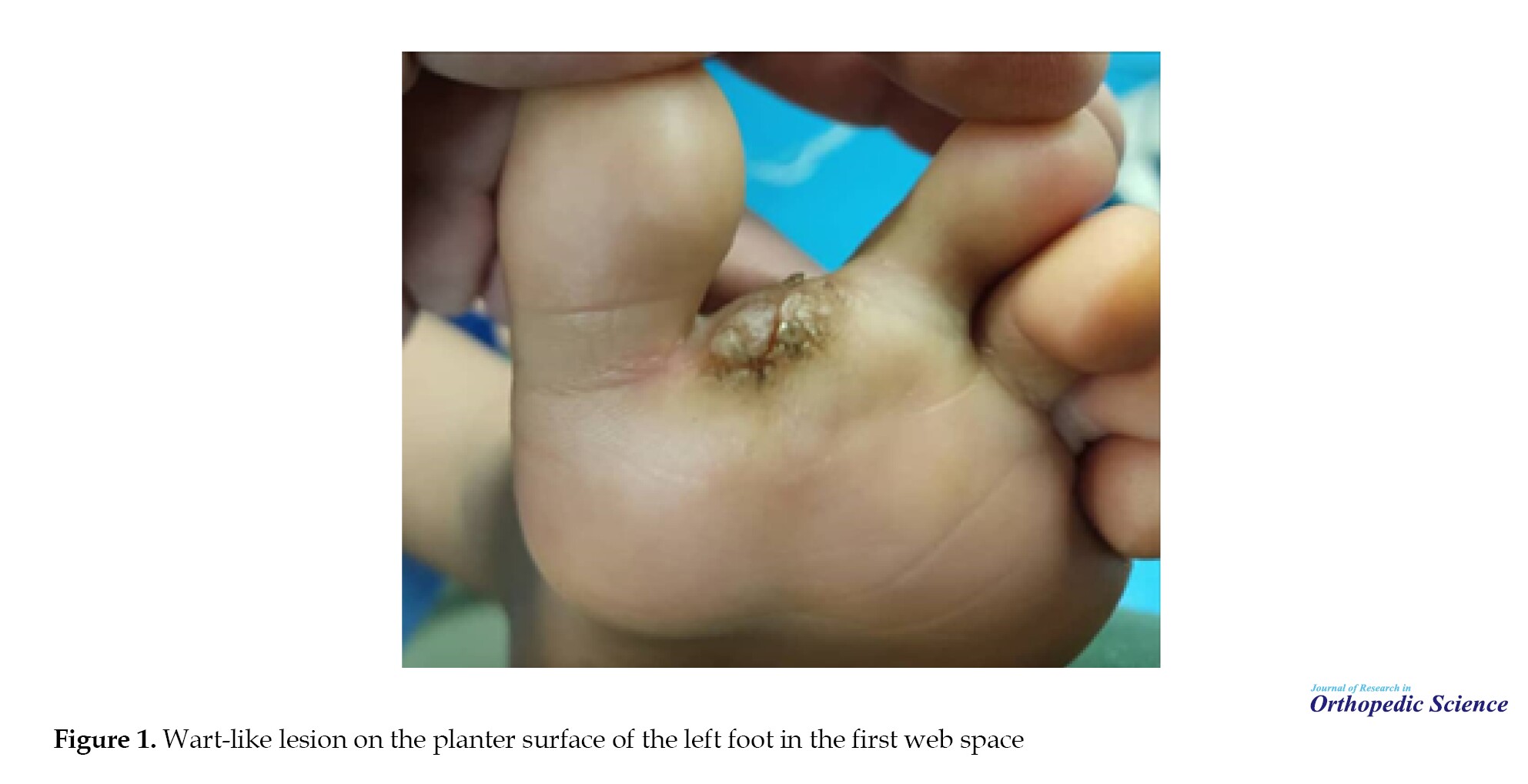
A 4-year-old Iranian boy was admitted to the Shafayahyaian Hospital, Iran, with pain and a wart-like lesion on the plantar surface of his left foot in the first web space over the past 4 months. The patient and his family reported no personal or family history of cancer, no comorbidities, no significant past medical history, no genetic issues, and no prior surgical history. Physical examination was unremarkable except for a plantar mass of the right foot between the first and second toes with mild pain, and the skin above the mass was softened (Figure 1).
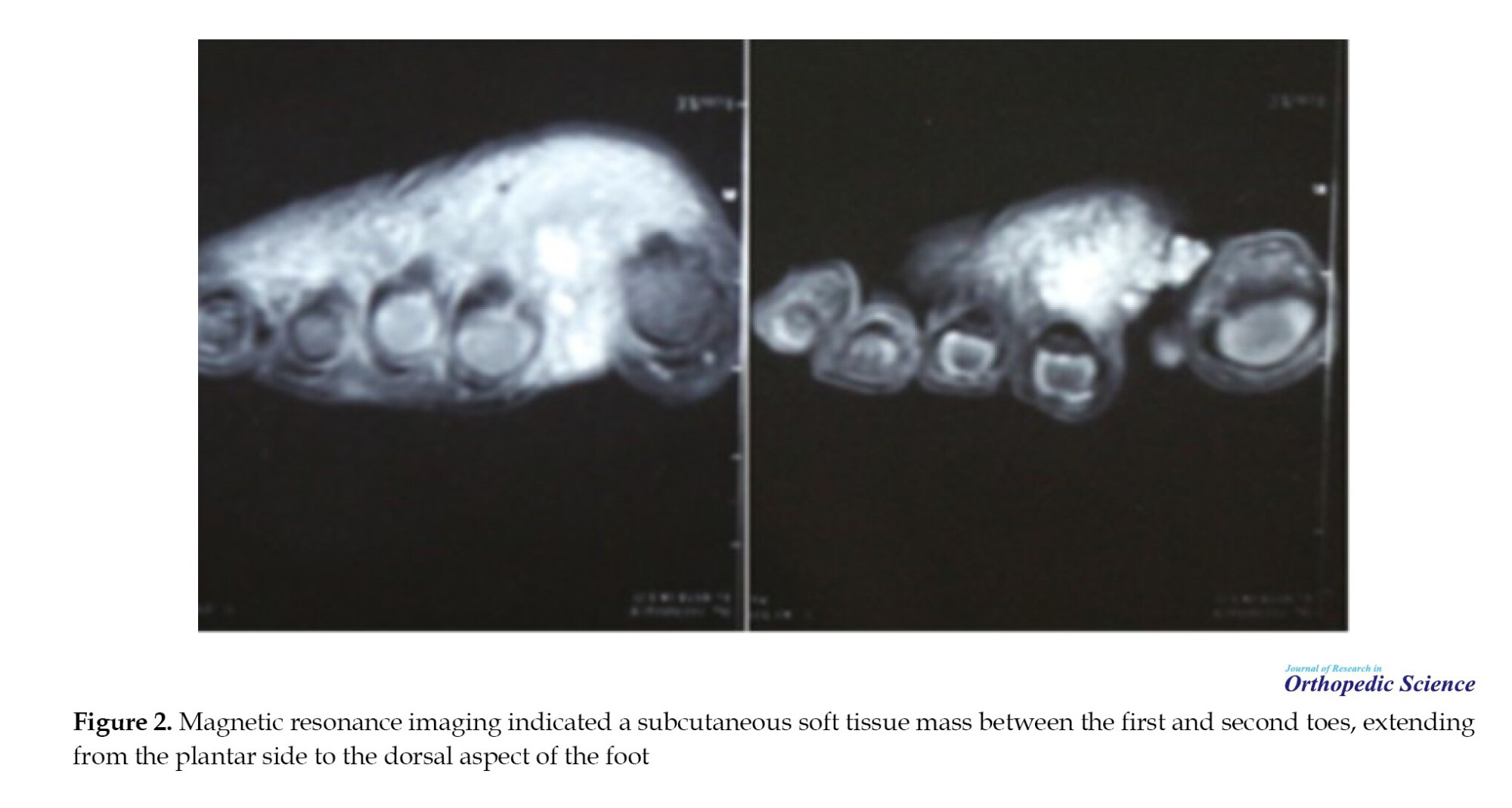
The patient’s laboratory tests were all within normal limits. Radiological workup with MRI revealed a 2×2 cm subcutaneous soft tissue mass in the area between the first and second toes, extending from the plantar side to the dorsal aspect of the foot (Figure 2). The results of the core-needle biopsy showed an infiltrative small blue round cell tumor suggestive of Ewing sarcoma. We scheduled the patient for excisional biopsy. The surgery was performed under general anesthesia with the application of a tourniquet to the proximal part of the lower extremity. After the surgery and following histology analysis, we confirmed this uncommon tumor.
Histopathology results:
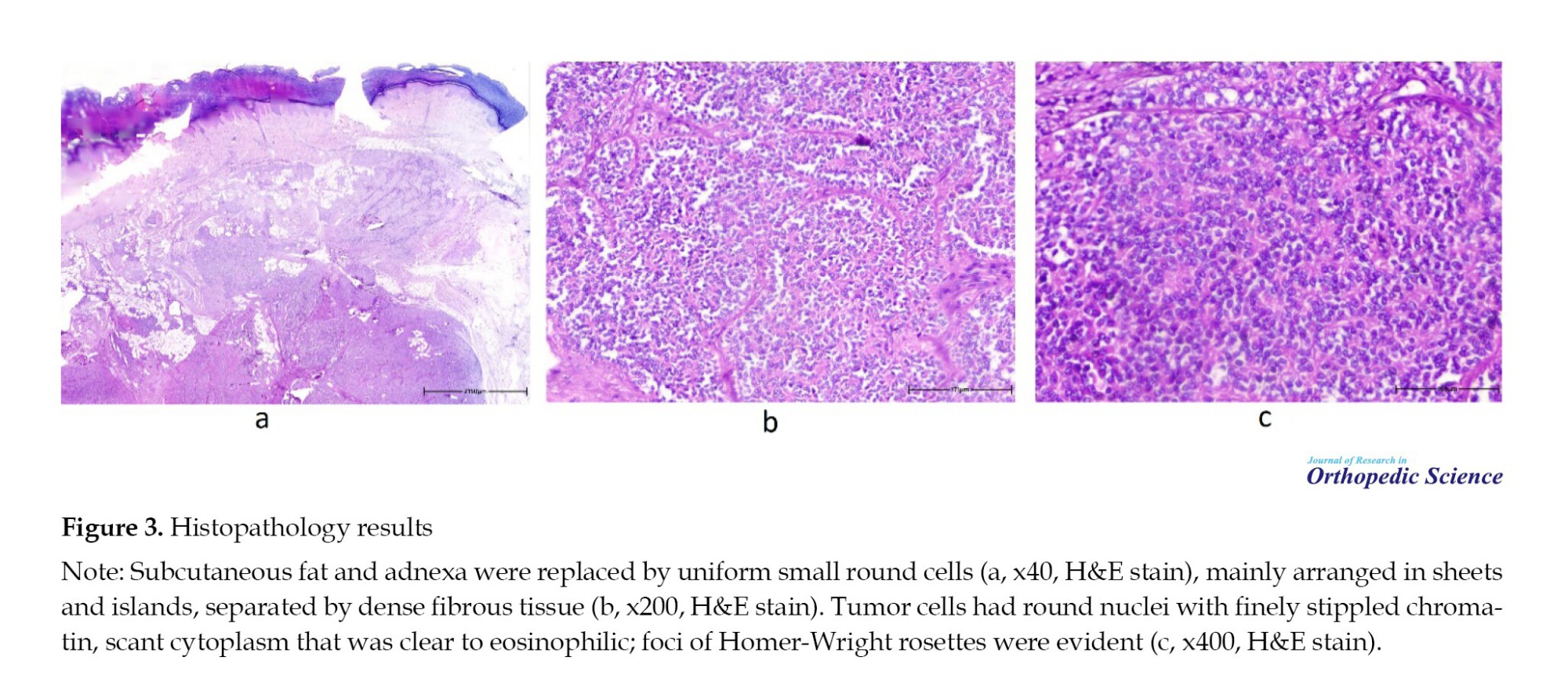
Histopathology sections showed skin extensively involved by uniform small round cells; destroying the adnexa and replacing subcutaneous fat without invasion of the epidermis. Tumor cells had round nuclei with finely stippled chromatin, inconspicuous nucleoli, and scant cytoplasm that was clear to eosinophilic with indistinct membranes; all were arranged in sheets and islands separated by dense fibrous tissue. Mitotic activity was brisk, and foci of Homer-Wright rosettes were evident. The neoplastic cells were strongly positive for FLI-1, showed diffuse membranous expression for CD99 and NSE, and were negative for WT-1, GFAP, and MyoD1 (Figures 3 and 4).

Discussion
ESFT is a group of tumors described by morphologically similar small, rounded cells and the presence of common histological and genetic features. This family consists of 4 types of tumors, including ES of the bone, pPNET, Askin tumor, and EES [2, 12, 13]. ES mostly affects bone tissue in children and adolescents. Although ES is typically considered a pediatric tumor, it can impact individuals up to their middle years [1]. The classification and diagnosis of these tumors is based on the histology and molecular evaluations [14, 15]. Although ES usually arises from the bone tissue, 20-30% of ESs originate from extraskeletal sites, known as EES [12, 16].
EES is a rare, aggressive tumor with an incidence of 0.1 to 0.4 per a million, 10 times lower than ES [17, 18]. Those with ESS are reported to be older than skeletal ES, with a bimodal distribution at <5 years and >35 years [16, 19]. Some of the most commonly reported sites for this extraskeletal sarcoma include the paravertebral region, chest wall, retroperitoneum, pelvis and hip, and the extremities [20]. EES symptoms are associated with the size and site of the tumor. In most cases, patients present with swelling and localized pain, in association with a rapidly growing mass [14]. Unfortunately, about 30% of cases show distant metastasis at the time of diagnosis [21].
Although patient outcomes in the two groups (EES and skeletal ES) have been reported to be similar when treated with ES protocols [5, 22, 23], more recent investigations documented better survival rate for EES [16, 24, 19]. Cash et al. investigated outcomes in patients with EES versus ES of the bone. They reported that although key tumor genomic features are the same, there are differences between the two groups regarding clinical characteristics. Prior studies also reported that there may be clinical differences between skeletal ES and EES [16, 24]. EES showed a better prognosis, independent of age, race, and primary site. They indicated that older age and an increased baseline erythrocyte sedimentation rate (ESR) are unfavorable prognostic factors in EES patients [19]. Other risk factors were reported as pelvic involvement, baseline leukocytosis, elevated Lactate dehydrogenase (LDH), and low hemoglobin (Hb) [22, 25]. The initial size of the tumor was also suggested as a strong prognostic factor in localized EES [26].
Imaging techniques, such as ultrasound, CT, and MRI, are used to diagnose and monitor the extent of the disease. MRI is the modality of choice among diagnostic imaging techniques and is used for local staging, followed by core-needle biopsy and pathological evaluation to reach a definitive diagnosis [2]. Pathological markers of the tumor are necessary for the definitive diagnosis of EES. In IHC, several markers, such as CD99 and FLI1 are studied [2]. According to NCCN and due to the rarity of EES and differences in its clinical presentation and in patients’ characteristics, there is no standard or definite guideline for the management of this condition [27, 28]. In EES cases occurring in children, this uncertainty is even greater, and more data from well-designed investigations are still needed [29]. EES management includes surgery and chemotherapy [30, 31]. Invasive management and surgical removal of superficial lesions in resectable tumors where negative margins are available can decrease the risk of metastatic spread and improve outcomes in patients. When tumors are unresectable, radiotherapy is the option of choice [32]. Long-term follow-up is essential to detect and manage potential complications in children with EES.
Our knowledge regarding the clinical features and incidence of EES in pediatrics is limited. A recent systematic review by Ghandour et al. on 5752 patients showed that 30% of the EES patients are children and adolescents. Interestingly, no association was reported between EES presentation and biological sex [29], consistent with previous investigations [16, 19]. This study highlighted that the thorax is the main origin of EES in children. The extremities, head and neck region, the pelvis, and the abdomen are among other common sites. However, few case reports describe some uncommon sites for EES, such as the great toe, frontal sinus, and kidneys [14, 11, 33]. A previous study reported a case of a 4-year-old patient with a solid, well-defined mass on the lateral plantar surface of his right great toe. Due to the increasing size of the tumor and onset of local pain, a surgical biopsy was performed, suggesting EES and confirmed by histological and IHC analysis [11].
Conclusion
EES is a rare malignant tumor with challenging diagnosis and management. Given the limited number of EES cases in the literature, there is no standard or definite guideline for its management. Although it is an uncommon site, it is important to consider this tumor in the differential diagnosis of cutaneous-subcutaneous lesions, especially in younger individuals or when a visible cutaneous lesion is present. These tumors are sometimes clinically misdiagnosed because they resemble other cutaneous cancers on examination. We reported a 4-year-old boy who was admitted with pain and a wart-like lesion on the plantar surface of his left foot, diagnosed as EES and confirmed by histopathology.
Ethical Considerations
Compliance with ethical guidelines
Ethical approval for this study was provided by Iran University of Medical Sciences, Tehran, Iran. The patient provided informed consent for the publication of this case report, which included the use of clinical images and details.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
Conceptualization and supervision: Babak Roshanravan and Khalil Kargar Shooroki; Methodology: Sam Hajialiloo Sami; Data collection: Amin HamidzadehKhiavi, Behnam Sour, and Mohammad Saleh Movahedi;Investigation and writing: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors gratefully acknowledge the contributions of all the patients who participated in this study, as well as the surgeons, nurses, and medical staff involved in surgery and treatment provided. Special thanks to the multidisciplinary teams whose collaboration made the comprehensive management of these complex cases possible.
References
Ewing sarcoma (ES), described by J. Ewing in 1921, is the second most common type of primary malignant bone tumor in children and young individuals. Although ES is typically considered a pediatric tumor, it can impact individuals up to the age of thirty [1]. It has been reported that about 25% of these aggressive tumors originate from soft tissue, known as extraskeletal ES (EES) [2]. EES, first documented in 1969 by Tefft et al., is an extremely uncommon entity that typically manifests in pediatrics and adolescents. Its incidence is reported to be about 1 in 5–10 million people, with most cases ranging between 10 and 20 years old [3, 4].
EES belongs to the ES family of tumors (ESFT), and its spherical malignant cells resemble one another in their molecular lineage [2, 5, 6]. According to available data, EES usually originates from the chest wall, proximal thigh, and extremities [7]. However, involvement of the breast, liver, and urinary tract has also been documented [8, 9]. The typical clinical manifestation of EES is an expanding lesion of about 2–3 cm, inducing compression symptoms, usually focal pain [7]. The mass is usually soft, movable, and superficial. Imaging studies, most importantly (MRI), are usually the first step in diagnosis, confirmed by histology and immunohistochemistry evaluations. Surgical resection, chemotherapy, and radiotherapy (in unresectable cases) are treatment options for EES management [2, 10].
Knowledge regarding the clinical features and incidence of EES in pediatric patients is limited. This study aimed to report an unusual case of EES of the foot in a 4-year-old Iranian boy, who was admitted with pain and a wart-like lesion on the plantar surface of his left foot in the first web space for the past 4 months. The diagnosis of subcutaneous EES in the foot is rare, with only a few cases reported at such a young age [11]. Although it is an uncommon site, it is important to consider EES, especially if there is a visible cutaneous lesion. The rarity of EES, along with relatively nonspecific findings in histology and immunophenotype, makes the diagnosis of superficial EES challenging, and radiologists, pathologists, and, more importantly, clinicians should consider these tumors in their differential diagnosis.
Case Presentation
A 4-year-old Iranian boy was admitted to the Shafayahyaian Hospital, Iran, with pain and a wart-like lesion on the plantar surface of his left foot in the first web space over the past 4 months. The patient and his family reported no personal or family history of cancer, no comorbidities, no significant past medical history, no genetic issues, and no prior surgical history. Physical examination was unremarkable except for a plantar mass of the right foot between the first and second toes with mild pain, and the skin above the mass was softened (Figure 1).
The patient’s laboratory tests were all within normal limits. Radiological workup with MRI revealed a 2×2 cm subcutaneous soft tissue mass in the area between the first and second toes, extending from the plantar side to the dorsal aspect of the foot (Figure 2). The results of the core-needle biopsy showed an infiltrative small blue round cell tumor suggestive of Ewing sarcoma. We scheduled the patient for excisional biopsy. The surgery was performed under general anesthesia with the application of a tourniquet to the proximal part of the lower extremity. After the surgery and following histology analysis, we confirmed this uncommon tumor.
Histopathology results:
Histopathology sections showed skin extensively involved by uniform small round cells; destroying the adnexa and replacing subcutaneous fat without invasion of the epidermis. Tumor cells had round nuclei with finely stippled chromatin, inconspicuous nucleoli, and scant cytoplasm that was clear to eosinophilic with indistinct membranes; all were arranged in sheets and islands separated by dense fibrous tissue. Mitotic activity was brisk, and foci of Homer-Wright rosettes were evident. The neoplastic cells were strongly positive for FLI-1, showed diffuse membranous expression for CD99 and NSE, and were negative for WT-1, GFAP, and MyoD1 (Figures 3 and 4).
Discussion
ESFT is a group of tumors described by morphologically similar small, rounded cells and the presence of common histological and genetic features. This family consists of 4 types of tumors, including ES of the bone, pPNET, Askin tumor, and EES [2, 12, 13]. ES mostly affects bone tissue in children and adolescents. Although ES is typically considered a pediatric tumor, it can impact individuals up to their middle years [1]. The classification and diagnosis of these tumors is based on the histology and molecular evaluations [14, 15]. Although ES usually arises from the bone tissue, 20-30% of ESs originate from extraskeletal sites, known as EES [12, 16].
EES is a rare, aggressive tumor with an incidence of 0.1 to 0.4 per a million, 10 times lower than ES [17, 18]. Those with ESS are reported to be older than skeletal ES, with a bimodal distribution at <5 years and >35 years [16, 19]. Some of the most commonly reported sites for this extraskeletal sarcoma include the paravertebral region, chest wall, retroperitoneum, pelvis and hip, and the extremities [20]. EES symptoms are associated with the size and site of the tumor. In most cases, patients present with swelling and localized pain, in association with a rapidly growing mass [14]. Unfortunately, about 30% of cases show distant metastasis at the time of diagnosis [21].
Although patient outcomes in the two groups (EES and skeletal ES) have been reported to be similar when treated with ES protocols [5, 22, 23], more recent investigations documented better survival rate for EES [16, 24, 19]. Cash et al. investigated outcomes in patients with EES versus ES of the bone. They reported that although key tumor genomic features are the same, there are differences between the two groups regarding clinical characteristics. Prior studies also reported that there may be clinical differences between skeletal ES and EES [16, 24]. EES showed a better prognosis, independent of age, race, and primary site. They indicated that older age and an increased baseline erythrocyte sedimentation rate (ESR) are unfavorable prognostic factors in EES patients [19]. Other risk factors were reported as pelvic involvement, baseline leukocytosis, elevated Lactate dehydrogenase (LDH), and low hemoglobin (Hb) [22, 25]. The initial size of the tumor was also suggested as a strong prognostic factor in localized EES [26].
Imaging techniques, such as ultrasound, CT, and MRI, are used to diagnose and monitor the extent of the disease. MRI is the modality of choice among diagnostic imaging techniques and is used for local staging, followed by core-needle biopsy and pathological evaluation to reach a definitive diagnosis [2]. Pathological markers of the tumor are necessary for the definitive diagnosis of EES. In IHC, several markers, such as CD99 and FLI1 are studied [2]. According to NCCN and due to the rarity of EES and differences in its clinical presentation and in patients’ characteristics, there is no standard or definite guideline for the management of this condition [27, 28]. In EES cases occurring in children, this uncertainty is even greater, and more data from well-designed investigations are still needed [29]. EES management includes surgery and chemotherapy [30, 31]. Invasive management and surgical removal of superficial lesions in resectable tumors where negative margins are available can decrease the risk of metastatic spread and improve outcomes in patients. When tumors are unresectable, radiotherapy is the option of choice [32]. Long-term follow-up is essential to detect and manage potential complications in children with EES.
Our knowledge regarding the clinical features and incidence of EES in pediatrics is limited. A recent systematic review by Ghandour et al. on 5752 patients showed that 30% of the EES patients are children and adolescents. Interestingly, no association was reported between EES presentation and biological sex [29], consistent with previous investigations [16, 19]. This study highlighted that the thorax is the main origin of EES in children. The extremities, head and neck region, the pelvis, and the abdomen are among other common sites. However, few case reports describe some uncommon sites for EES, such as the great toe, frontal sinus, and kidneys [14, 11, 33]. A previous study reported a case of a 4-year-old patient with a solid, well-defined mass on the lateral plantar surface of his right great toe. Due to the increasing size of the tumor and onset of local pain, a surgical biopsy was performed, suggesting EES and confirmed by histological and IHC analysis [11].
Conclusion
EES is a rare malignant tumor with challenging diagnosis and management. Given the limited number of EES cases in the literature, there is no standard or definite guideline for its management. Although it is an uncommon site, it is important to consider this tumor in the differential diagnosis of cutaneous-subcutaneous lesions, especially in younger individuals or when a visible cutaneous lesion is present. These tumors are sometimes clinically misdiagnosed because they resemble other cutaneous cancers on examination. We reported a 4-year-old boy who was admitted with pain and a wart-like lesion on the plantar surface of his left foot, diagnosed as EES and confirmed by histopathology.
Ethical Considerations
Compliance with ethical guidelines
Ethical approval for this study was provided by Iran University of Medical Sciences, Tehran, Iran. The patient provided informed consent for the publication of this case report, which included the use of clinical images and details.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
Conceptualization and supervision: Babak Roshanravan and Khalil Kargar Shooroki; Methodology: Sam Hajialiloo Sami; Data collection: Amin HamidzadehKhiavi, Behnam Sour, and Mohammad Saleh Movahedi;Investigation and writing: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgments
The authors gratefully acknowledge the contributions of all the patients who participated in this study, as well as the surgeons, nurses, and medical staff involved in surgery and treatment provided. Special thanks to the multidisciplinary teams whose collaboration made the comprehensive management of these complex cases possible.
References
- Lahl M, Fisher VL, Laschinger K. Ewing's sarcoma family of tumors: An overview from diagnosis to survivorship. Clin J Oncol Nurs. 2008; 12(1):89-97. [DOI:10.1188/08.CJON.89-97] [PMID]
- Abboud A, Masrouha K, Saliba M, Haidar R, Saab R, Khoury N, et al. Extraskeletal Ewing sarcoma: Diagnosis, management and prognosis. Oncol Lett. 2021; 21(5):354.[DOI:10.3892/ol.2021.12615] [PMID]
- Jiang S, Wang G, Chen J, Dong Y. Comparison of clinical features and outcomes in patients with extraskeletal vs skeletal Ewing sarcoma: An SEER database analysis of 3,178 cases. Cancer Manag Res. 2018; 10:6227-36. [DOI:10.2147/CMAR.S178979] [PMID]
- Cote GM, Choy E. Update in treatment and targets in Ewing sarcoma. Hematol Oncol Clin North Am. 2013; 27(5):1007-19. [DOI:10.1016/j.hoc.2013.07.001] [PMID]
- El Weshi A, Allam A, Ajarim D, Al Dayel F, Pant R, Bazarbashi S, et al. Extraskeletal Ewing’s sarcoma family of tumours in adults: Analysis of 57 patients from a single institution. Clin Oncol (R Coll Radiol). 2010; 22(5):374-81. [DOI:10.1016/j.clon.2010.02.010] [PMID]
- Rud NP, Reiman HM, Pritchard DJ, Frassica FJ, Smithson WA. Extraosseous Ewing’s sarcoma. A study of 42 cases. Cancer. 1989; 64(7):1548-53. [DOI:10.1002/1097-0142(19891001)64:73.0.CO;2-W] [PMID]
- Weiss SW, Goldblum JR, Folpe AL. Enzinger and Weiss’s soft tissue tumors. Amsterdam: Elsevier Health Sciences; 2007. [Link]
- Srivastava S, Arora J, Parakh A, Goel RK. Primary extraskeletal Ewing’s sarcoma/primitive neuroectodermal tumor of breast. Indian J Radiol Imaging. 2016; 26(2):226-30. [DOI:10.4103/0971-3026.184408] [PMID]
- Aboumarzouk OM, Coleman R, Goepel JR, Shorthouse AJ. PNET/Ewing’s sarcoma of the rectum: A case report and review of the literature. Case Reports. 2009; 2009:bcr0420091770. [DOI:10.1136/bcr.04.2009.1770] [PMID]
- Wright A, Desai M, Bolan CW, Badawy M, Guccione J, Rao Korivi B, et al. Extraskeletal Ewing sarcoma from head to toe: Multimodality imaging review. Radiographics. 2022; 42(4):1145-60. [DOI:10.1148/rg.210226] [PMID]
- Cypel TK, Meilik B, Zuker RM. Extraskeletal Ewing’s sarcoma in a great toe of a young boy. Can J Plast Surg. 2007; 15(3):165-8. [DOI:10.1177/229255030701500306] [PMID]
- Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010; 11(2):184-92. [DOI:10.1016/S1470-2045(09)70286-4] [PMID]
- Koscielniak E, Sparber‐Sauer M, Scheer M, Vokuhl C, Kazanowska B, Ladenstein R, et al. Extraskeletal Ewing sarcoma in children, adolescents, and young adults. An analysis of three prospective studies of the cooperative weichteilsarkomstudiengruppe (CWS). Pediatr Blood Cancer. 2021; 68(10):e29145. [DOI:10.1002/pbc.29145] [PMID]
- Costa IE, Menezes AS, Lima AF, Rodrigues B. Extra-skeletal Ewing’s sarcoma of the frontal sinus: A rare disorder in pediatric age. BMJ Case Rep. 2020; 13(5):e232460.[DOI:10.1136/bcr-2019-232460] [PMID]
- Kim SK, Park YK. Ewing sarcoma: a chronicle of molecular pathogenesis. Hum Pathol. 2016; 55:91-100. [DOI:10.1016/j.humpath.2016.05.008] [PMID]
- Applebaum MA, Worch J, Matthay KK, Goldsby R, Neuhaus J, West DC, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 2011; 117(13):3027-32. [DOI:10.1002/cncr.25840] [PMID]
- Shrateh ON, Jobran AWM, Owienah H, Sweileh T, Abulihya M, Natsheh MA, et al. Primary extraskeletal Ewing sarcoma of the foot with extensive skeletal and pulmonary metastasis: A rare case report. Ann Med Surg (Lond). 2022; 82:104752. [DOI:10.1016/j.amsu.2022.104752] [PMID]
- Zegeye AM, Alemayehu BT, Worku DT, Abera KA, Abera SA, Alemu BA. Extraskeletal Ewing’s sarcoma of the thigh with lung and bone metastasis: Case report. Int J Surg Case Rep. 2023; 112:108919. [DOI:10.1016/j.ijscr.2023.108919] [PMID]
- Cash T, McIlvaine E, Krailo MD, Lessnick SL, Lawlor ER, Laack N, et al. Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: a report from the children’s oncology group. Pediatr Blood Cancer. 2016; 63(10):1771-9. [DOI:10.1002/pbc.26096] [PMID]
- Javalgi AP, Karigoudar MH, Palur K. Blue cell tumour at unusual site: Retropritoneal Ewings sarcoma. J Clin Diagn Res. 2016; 10(4):ED19-20. [DOI:10.7860/JCDR/2016/18302.7618] [PMID]
- Ansari MH, Gujrathi AB, Ambulgekar V. Extraskeletal Ewing's sarcoma of neck in a child- a case report. Iran J Otorhinolaryngol. 2019; 31(104):173-6. [PMID]
- Lee JA, Kim DH, Lim JS, Koh JS, Kim MS, Kong CB, et al. Soft-tissue Ewing sarcoma in a low-incidence population: comparison to skeletal Ewing sarcoma for clinical characteristics and treatment outcome. Jpn J Clin Oncol. 2010; 40(11):1060-7. [DOI:10.1093/jjco/hyq080] [PMID]
- Castex MP, Rubie H, Stevens MC, Escribano CC, de Gauzy JS, Gomez-Brouchet A, et al. Extraosseous localized ewing tumors: improved outcome with anthracyclines--the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol. 2007; 25(10):1176-82. [DOI:10.1200/JCO.2005.05.0559] [PMID]
- Biswas B, Shukla NK, Deo SV, Agarwala S, Sharma DN, Vishnubhatla S, et al. Evaluation of outcome and prognostic factors in extraosseous Ewing sarcoma. Pediatr Blood Cancer. 2014; 61(11):1925-31 [DOI:10.1002/pbc.25095] [PMID]
- Orr WS, Denbo JW, Billups CA, Wu J, Navid F, Rao BN, et al. Analysis of prognostic factors in extraosseous Ewing sarcoma family of tumors: Review of St. Jude Children's Research Hospital experience. Ann Surg Oncol. 2012; 19(12):3816-22. [DOI:10.1245/s10434-012-2458-4] [PMID]
- Brinkhuis M, Wijnaendts L, Van der Linden J, Baak J, Meijer C, van Unnik A, et al. Peripheral primitive neuroectodermal tumour and extra-osseous Ewing’s sarcoma; A histological, immunohistochemical and DNA flow cytometric study. Virchows Arch. 1995; 425(6):611-6. [DOI:10.1007/BF00199351] [PMID]
- Casali PG, Bielack S, Abecassis N, Aro H, Bauer S, Biagini R, et al. Bone sarcomas: ESMO-PaedCan-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; 29(Suppl 4):iv79-iv95. [PMID]
- Biermann JS. Updates in the treatment of bone cancer. J Natl Compr Canc Netw. 2013; 11(5 Suppl):681-3. [DOI:10.6004/jnccn.2013.0200] [PMID]
- Ghandour M, Lehner B, Klotz M, Geisbuesch A, Bollmann J, Renkawitz T, et al. Extraosseous Ewing sarcoma in children: A systematic review and meta-analysis of clinicodemographic characteristics. Children. 2022; 9(12):1859. [DOI:10.3390/children9121859] [PMID]
- Mori Y, Kinoshita S, Kanamori T, Kataoka H, Joh T, Iida S, et al. The successful treatment of metastatic extraosseous Ewing sarcoma with pazopanib. Intern Med. 2018; 57(18):2753-7. [DOI:10.2169/internalmedicine.9879-17] [PMID]
- Bailey K, Cost C, Davis I, Glade-Bender J, Grohar P, Houghton P, et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the children’s oncology group (COG) New Agents for Ewing sarcoma task force. F1000Res. 2019; 8:F1000 Faculty Rev-493. [DOI:10.12688/f1000research.18139.1] [PMID]
- Momin FJ, Bhattacharyya M, Saikia S, Mishra RK, Nath J, Sarma G, et al. Radiotherapy in Ewing’s sarcoma family tumor-experience from north-east India. Oncol Clin Pract. 2021; 17(3):103-11. [DOI:10.5603/OCP.2021.0008]
- Tarek N, Said R, Andersen CR, Suki TS, Foglesong J, Herzog CE, et al. Primary Ewing sarcoma/primitive neuroectodermal tumor of the kidney: The MD Anderson Cancer center experience. Cancers (Basel). 2020; 12(10):2927. [DOI:10.3390/cancers12102927] [PMID]
Type of Study: Case Report |
Subject:
Tumor surgery
Received: 2025/02/11 | Accepted: 2025/03/16 | Published: 2025/05/1
Received: 2025/02/11 | Accepted: 2025/03/16 | Published: 2025/05/1
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |

Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
